Creating a Variant-Modified Reference
Stevie Pederson
Black Ochre Data Labs, The Kids Research Institute Australia, Adelaide, Australiastevie.pederson@thekids.org.au Source:
vignettes/creating_a_new_reference.Rmd
creating_a_new_reference.RmdAbstract
The use of personalised or population-level variants opens the door to the possibility of creating a variant-modified reference genome, or transcriptome. The package transmogR allows the creation of both, with a focus on combining both in order to created a custom reference transcriptome, along with decoy transcripts for use with the pseudo-aligner salmon.
Introduction
The incorporation of personalised or population-level variants into a
reference genome has been shown to have a significant impact on
subsequent alignments (Kaminow et al.
2022). Whilst implemented for splice-aware alignment or RNA-Seq
data using STARconsensus, the package transmogR
enables the creation of a variant-modified reference
transcriptome for use with pseudo aligners such as salmon
(Srivastava et al. 2020). In addition,
multiple visualisation and summarisation methods are included for a
cursory analysis of any custom variant sets being used.
Whilst the subsequent code is demonstrated on a small genomic region, the complete process for creating a modified a reference can run in under 20 minutes if using 4 or more cores.
Importantly, it is expected that the user will have carefully prepared their set of variants such that only the following exact variant types are included.
- SNV/SNP: The associated range must be of width 1 with a single base as the replacement allele
- Insertions: The position being replaced must be a single nucleotide with the replacement bases being > 1nt
- Deletions: The position being replaced must be > 1nt, whilst the replacement allele must be a single base
Any variants which do not conform to these criteria will likely cause a series of frustrating errors when attempting to use this package.
Setup
Installation
In order to perform the operations in this vignette, the following packages require installation.
if (!"BiocManager" %in% rownames(installed.packages()))
install.packages("BiocManager")
BiocManager::install("transmogR")Once these packages are installed, we can load them easily
library(VariantAnnotation)
library(rtracklayer)
library(extraChIPs)
library(transmogR)
library(GenomicFeatures)
library(BSgenome.Hsapiens.UCSC.hg38)Required Data
In order to create a modified reference, three primary data objects are required: 1) a reference genome; 2) a set of genomic variants; and 3) a set of exon-level co-ordinates defining transcript structure.
For this vignette, we’ll use GRCh38 as our primary reference genome,
but restricting the sequences to chr1 only. The package can
take either a DNAStringSet or BSgenome object
as the reference genome.
chr1 <- getSeq(BSgenome.Hsapiens.UCSC.hg38, "chr1")
chr1 <- as(chr1, "DNAStringSet")
names(chr1) <- "chr1"
chr1## DNAStringSet object of length 1:
## width seq names
## [1] 248956422 NNNNNNNNNNNNNNNNNNNNN...NNNNNNNNNNNNNNNNNNNNN chr1Given this represents the complete reference genome, we can also
setup a Seqinfo object for all downstream analysis, and to
ensure all objects are working with the same reference genome.
## Seqinfo object with 1 sequence from GRCh38 genome:
## seqnames seqlengths isCircular genome
## chr1 248956422 NA GRCh38A small set of variants from the 1000 Genomes Project1 has been provided with the package in VCF format.
vcf <- system.file("extdata/1000GP_subset.vcf.gz", package = "transmogR")
vcf <- VcfFile(vcf)Variant callers can often produce variant calls which are
incompatible and the function checkVariants() will check
for overlapping InDels, as well as returning both the REF
and ALT columns as a character vectors. This function can
be applied to any set of variants parsed by other tools, or can be
applied directly to a VcfFile to check variants whilst
parsing.
var <- cleanVariants(vcf)
seqinfo(var) <- sq
var## GRanges object with 100 ranges and 2 metadata columns:
## seqnames ranges strand | REF
## <Rle> <IRanges> <Rle> | <character>
## 1:113969:C:T chr1 113969 * | C
## 1:121009:C:T chr1 121009 * | C
## 1:123511:G:A chr1 123511 * | G
## 1:126113:C:A chr1 126113 * | C
## 1:128798:C:T chr1 128798 * | C
## ... ... ... ... . ...
## 1:839405:G:A chr1 839405 * | G
## 1:839480:CACACACCTG:C chr1 839480-839494 * | CACACACCTGGACAA
## 1:839515:CTAGACACAC:C chr1 839515-839543 * | CTAGACACACACACCTGGAC..
## 1:840046:G:A chr1 840046 * | G
## 1:840279:A:G chr1 840279 * | A
## ALT
## <character>
## 1:113969:C:T T
## 1:121009:C:T T
## 1:123511:G:A A
## 1:126113:C:A A
## 1:128798:C:T T
## ... ...
## 1:839405:G:A A
## 1:839480:CACACACCTG:C C
## 1:839515:CTAGACACAC:C C
## 1:840046:G:A A
## 1:840279:A:G G
## -------
## seqinfo: 1 sequence from GRCh38 genomeIn this case, no incompatible variants were included, but if they are
present, an error will be produced by default. This behaviour can be
modified by setting the ol_vars argument to return no
overlapping InDels (ol_vars = 'none'), the longest or
shortest in terms of the largest or smallest change to the chromosome
length, or even the first or last of the overlapping variants.
An additional set of transcripts derived from Gencode v442 has also been provided.
f <- system.file("extdata/gencode.v44.subset.gtf.gz", package = "transmogR")
gtf <- import.gff(f, which = GRanges(sq))
seqinfo(gtf) <- sq
gtf## GRanges object with 603 ranges and 11 metadata columns:
## seqnames ranges strand | source type score
## <Rle> <IRanges> <Rle> | <factor> <factor> <numeric>
## [1] chr1 89295-133723 - | rtracklayer gene NA
## [2] chr1 89295-120932 - | rtracklayer transcript NA
## [3] chr1 120775-120932 - | rtracklayer exon NA
## [4] chr1 112700-112804 - | rtracklayer exon NA
## [5] chr1 92091-92240 - | rtracklayer exon NA
## ... ... ... ... . ... ... ...
## [599] chr1 852671-852766 + | rtracklayer exon NA
## [600] chr1 853391-854096 + | rtracklayer exon NA
## [601] chr1 851348-852752 + | rtracklayer transcript NA
## [602] chr1 851348-852110 + | rtracklayer exon NA
## [603] chr1 852671-852752 + | rtracklayer exon NA
## phase gene_id gene_type gene_name
## <integer> <character> <character> <character>
## [1] <NA> ENSG00000238009.6 lncRNA ENSG00000238009
## [2] <NA> ENSG00000238009.6 lncRNA ENSG00000238009
## [3] <NA> ENSG00000238009.6 lncRNA ENSG00000238009
## [4] <NA> ENSG00000238009.6 lncRNA ENSG00000238009
## [5] <NA> ENSG00000238009.6 lncRNA ENSG00000238009
## ... ... ... ... ...
## [599] <NA> ENSG00000228794.12 lncRNA LINC01128
## [600] <NA> ENSG00000228794.12 lncRNA LINC01128
## [601] <NA> ENSG00000228794.12 lncRNA LINC01128
## [602] <NA> ENSG00000228794.12 lncRNA LINC01128
## [603] <NA> ENSG00000228794.12 lncRNA LINC01128
## transcript_id transcript_type transcript_name exon_number
## <character> <character> <character> <character>
## [1] <NA> <NA> <NA> <NA>
## [2] ENST00000466430.5 lncRNA ENST00000466430 <NA>
## [3] ENST00000466430.5 lncRNA ENST00000466430 1
## [4] ENST00000466430.5 lncRNA ENST00000466430 2
## [5] ENST00000466430.5 lncRNA ENST00000466430 3
## ... ... ... ... ...
## [599] ENST00000688420.1 lncRNA LINC01128-235 4
## [600] ENST00000688420.1 lncRNA LINC01128-235 5
## [601] ENST00000425657.1 retained_intron LINC01128-203 <NA>
## [602] ENST00000425657.1 retained_intron LINC01128-203 1
## [603] ENST00000425657.1 retained_intron LINC01128-203 2
## -------
## seqinfo: 1 sequence from GRCh38 genomeSplitting this gtf into feature types can also be very handy for downstream processes.
gtf <- splitAsList(gtf, gtf$type)Inspecting Variants
Knowing where our variants lie, and how they relate to each other can be informative, and as such, some simple summarisation functions have been included. Knowing where our variants lie, and how they relate to each other can be informative, and as such, some simple visualisation and summarisation functions have been included. In the following, we can check to see how many exons directly overlap a variant, showing how many unique genes this summarises to. Any ids, which don’t overlap a variants are also described in the plot title.
upsetVarByCol(gtf$exon, var, mcol = "gene_id")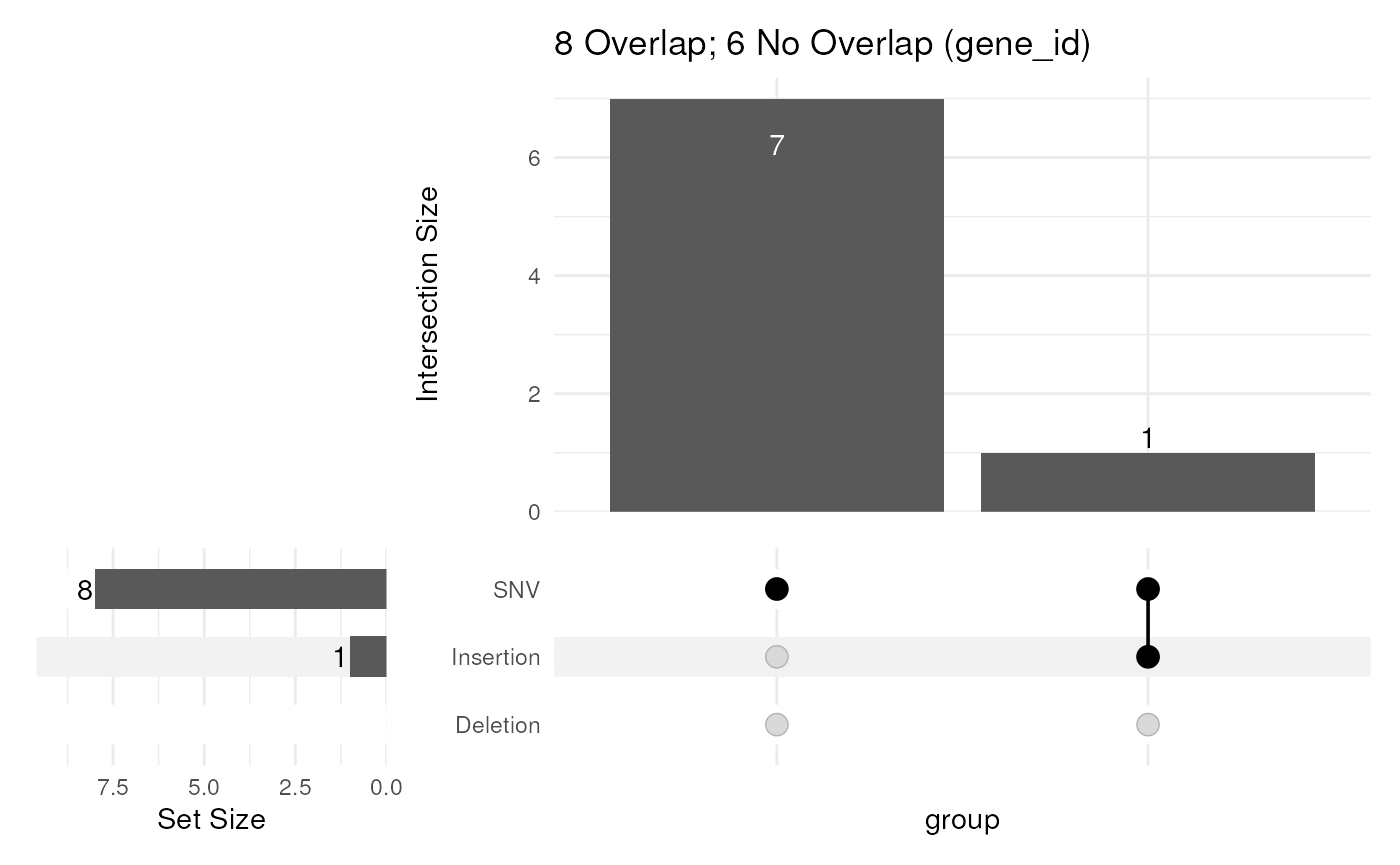
Included variants which overlap exonic sequences, summarised by unique gene ids
In addition, we can obtain a simple breakdown of overlapping regions
using a GRangesList. We can obtain a simple breakdown of overlapping
regions using a GRangesList. We can use the function
defineRegions() from extraChIPs to define
regions based on gene & transcript locations. This function assigns
each position in the genome uniquely to a given feature hierarchically,
using all provided transcripts, meaning some exons will be considered as
promoters. To ensure that all exons are included as exons, we can just
substitute in the values from our gtf for this feature type.
regions <- defineRegions(gtf$gene, gtf$transcript, gtf$exon, proximal = 0)
regions$exon <- granges(gtf$exon)
overlapsByVar(regions, var)## Deletion Insertion SNV Total
## promoter 1 1 46 48
## upstream_promoter 0 0 20 20
## exon 0 1 37 38
## intron 5 0 24 29
## intergenic 0 0 0 0Creating Modified Reference Sequences
Several approaches are available for creating modified reference genomes or transcriptomes, as outlines below.
Modifying a Reference Transcriptome
A simple option for creating a variant-modified reference
transcriptome is to call the function transmogrify(),
providing the reference sequence, a set of variants and the genomic
co-ordinates of sequence features (e.g. transcripts) for which the
sequences are required.
An optional tag can be added to all transcripts to indicate which
have been modified, with an additional tag able to be included which
indicates which type of variant has been incorporated into the new
transcript sequence. Variant tags will be one of s,
i or d to indicate SNV, Insertions or
Deletions respectively. In our example dataset, only one transcript
contains both SNVs and an insertion.
trans_mod <- transmogrify(chr1, var, gtf$exon, tag = "1000GP", var_tags = TRUE)
trans_mod## DNAStringSet object of length 112:
## width seq names
## [1] 1947 ACCCTCCTTGAGACAGCCCTCC...TAAACAATACACACGTGTTAAA ENST00000326734.2...
## [2] 1702 CACACCGTGAGCTGCTGAGACG...GTGCAGGGCACAGGTGGGCGCC ENST00000357876.6
## [3] 1358 AATCAGAACTCGCGGTGGGGGC...ATAAAATTAATGAGAATGATCT ENST00000412115.2...
## [4] 421 GACAGGGTCTCCCTCTGTTGTC...AAAGCATCCAGGATTCAATGAA ENST00000414688.6
## [5] 894 CTGCAATACCTCACTCAATCTC...GTATTTGATGGCCTCTGTTGTT ENST00000415295.1
## ... ... ...
## [108] 3097 GCCAGTGTCTCCGCCGGTTGAA...GATAAAACTTAAAAATATCCCC ENST00000701768.1
## [109] 1601 GGAGTGCGTTCGGTGCGCCGTG...AGCTATTAAAAGAGACAGAGGC ENST00000702098.1
## [110] 1432 GTCTGCGTCGGGTTCTGTTGGA...ATCAATAAAAATTTAAATGCTC ENST00000702273.1
## [111] 1460 CTGTTAGGTAAGAGGCGCGTGA...ATCAATAAAAATTTAAATGCTC ENST00000702557.1
## [112] 1651 GTTAGGTAAGAGGCGCGTGAGG...GCTATTAAAAGAGACAGAGGCA ENST00000702847.1## [1] "ENST00000473798.2_1000GP_si"This can be simply exported again using
writeXStringSet().
This approach effectively steps through all the provided transcripts inserting the relevant variants where required, and as such can be somewhat time-consuming when working with genome-wide variants and a complete transcriptome.
Modifying a Reference Genome
In addition to creating a set of modified transcripts, the genomic
sequence can also be modified with a call to genomogrify(),
using either a DNAStringSet or BSgenome object
to provide the reference sequences. A tag can be optionally added to all
sequence names to avoid confusion, however, in the below example this
option has not been utilised
chr1_mod <- genomogrify(chr1, var)
chr1_mod## DNAStringSet object of length 1:
## width seq names
## [1] 248956362 NNNNNNNNNNNNNNNNNNNNN...NNNNNNNNNNNNNNNNNNNNN chr1The new reference genome can be exported to fasta format using
writeXStringSet(). Incorporation of both of these into a
‘gentrome’ for use with the pseudo-aligner salmon (Srivastava et al. 2020) is a straight-forward
concatenation.
c(trans_mod, chr1_mod) %>% writeXStringSet("gentrome.fa.gz", compress = TRUE)
writeLines(names(chr1_mod), "decoys.txt")Finding Modified Co-ordinates
Both genomogrify() and transmogrify() rely
on the lower-level functions owl() and
indelcator() which overwrite letters or substitute
indels respectively. These are also able to be called
individually as shown in the help pages. However,
transmogrify() can run for an extended time if modifying
tens of thousands of transcripts, as would be the case for creating an
entirely new reference transcriptome.
In addition to the above, the co-ordinates from a GRanges object can
be modified to account for all length-modifying variants, i.e. InDels.
These shifted co-ordinates can then be used with a variant-modified
reference genome to directly extract the modified transcript sequences
using extractTranscriptSeq() from
GenomicFeatures (Lawrence et al.
2013). This is traditionally performed using a
GRangesList with each element containing the exons for a
given transcript.
ref_exons_by_trans <- gtf$exon %>% splitAsList(.$transcript_id)
ref_trans <- extractTranscriptSeqs(chr1, ref_exons_by_trans)
ref_trans## DNAStringSet object of length 112:
## width seq names
## [1] 1947 ACCCTCCTTGAGACAGCCCTCC...TAAACAATACACACGTGTTAAA ENST00000326734.2
## [2] 1702 CACACCGTGAGCTGCTGAGACG...GTGCAGGGCACAGGTGGGCGCC ENST00000357876.6
## [3] 1358 AATCAGAACTCGCGGTGGGGGC...ATAAAATTAATGAGAATGATCT ENST00000412115.2
## [4] 421 GACAGGGTCTCCCTCTGTTGTC...AAAGCATCCAGGATTCAATGAA ENST00000414688.6
## [5] 894 CTGCAATACCTCACTCAATCTC...GTATTTGATGGCCTCTGTTGTT ENST00000415295.1
## ... ... ...
## [108] 3097 GCCAGTGTCTCCGCCGGTTGAA...GATAAAACTTAAAAATATCCCC ENST00000701768.1
## [109] 1601 GGAGTGCGTTCGGTGCGCCGTG...AGCTATTAAAAGAGACAGAGGC ENST00000702098.1
## [110] 1432 GTCTGCGTCGGGTTCTGTTGGA...ATCAATAAAAATTTAAATGCTC ENST00000702273.1
## [111] 1460 CTGTTAGGTAAGAGGCGCGTGA...ATCAATAAAAATTTAAATGCTC ENST00000702557.1
## [112] 1651 GTTAGGTAAGAGGCGCGTGAGG...GCTATTAAAAGAGACAGAGGCA ENST00000702847.1In order to perform an analogous operation on a variant-modified
reference, we need to shift the co-ordinates of any feature in
accordance with the variants incoporated into the modified reference.
Whilst not being a concern if only SNVs are used as variants, any
Insertion or Deletion will shift genomic co-ordinates. This can be
performed using shiftByVar() which returns a set of GRanges
with co-ordinates shifted to account for any variants
new_exons <- shiftByVar(gtf$exon, var)This will return an object with a modified Seqinfo()
object so that sequence lengths will match the modified chromosomes.
seqinfo(new_exons)## Seqinfo object with 1 sequence from GRCh38 genome:
## seqnames seqlengths isCircular genome
## chr1 248956362 NA GRCh38
seqinfo(chr1_mod)## Seqinfo object with 1 sequence from an unspecified genome:
## seqnames seqlengths isCircular genome
## chr1 248956362 NA <NA>This enables simple extraction of transcript sequences using the
capabilities of GenomicFeatures
new_exon_by_trans <- splitAsList(new_exons, new_exons$transcript_id)Whilst transmogrify will add tags to transcript names
when requested, if choosing this approach, tags may be added manually,
with varTags() being designed for this possbility.
Importantly, checking for overlaps with variants needs to be performed
before shifting the co-ordinates, and these can then be added
at any subsequent point in the process where the order of the transcript
names is known.
tags <- varTags(ref_exons_by_trans, var, tag = "1000GP")
names(new_exon_by_trans) <- paste0(names(new_exon_by_trans), tags)Now, the transcript sequences can be extracted, with tags incorporated
new_trans_mod <- extractTranscriptSeqs(chr1_mod, new_exon_by_trans)This will return all unmodified transcripts, as unmodified, despite
the new co-ordinates, and will return modified transcripts which are
identical to those returned by transmogrify()
## [1] TRUE
all(new_trans_mod[modified] == trans_mod[modified])## [1] TRUEIn general, this approach is much faster and less resource hungry
than using transmogrify(), however both approaches will
require significant RAM. Whilst some common-use laptops may have the
required capacity, resources provided by HPC access will guarantee the
process will execute to completion.
Additional Capabilities
Pseudo-Autosomal Regions (PAR-Y)
Beyond these lower-level functions, PAR-Y regions for hg38, hg19 and
CHM13 are able to obtained using parY() and passing the
appropriate seqinfo object. This seqinfo
object checks the length of the Y-chromosome and guesses which reference
genome has been used.
## GRanges object with 2 ranges and 1 metadata column:
## seqnames ranges strand | PAR
## <Rle> <IRanges> <Rle> | <character>
## [1] chrY 10001-2781479 * | PAR1
## [2] chrY 56887903-57217415 * | PAR2
## -------
## seqinfo: 711 sequences (1 circular) from hg38 genomeIf the user wishes to exclude transcripts in the PAR-Y region, these
ranges can be passed to transmogrify() and any transcripts
which overlap the PAR-Y region will be excluded. Alternatively, passing
the entire Y-chromosome to transmogrify() will exclude all
transcripts in the Y-chromosome, as may be preferred for female-specific
references.
These regions can also be passed to genomogrify() as a
mask, with all bases within the masked region being re-assigned as an
N.
Splice Junctions
In addition, the set of splice junctions associated with any transcript can be obtained using our known exons.
ec <- c("transcript_id", "transcript_name", "gene_id", "gene_name")
sj <- sjFromExons(gtf$exon, extra_cols = ec)
sj## GRanges object with 730 ranges and 6 metadata columns:
## seqnames ranges strand | site transcript_id
## <Rle> <IRanges> <Rle> | <character> <character>
## [1] chr1 182745-182752 + | donor ENST00000624431.2
## [2] chr1 183128-183132 + | acceptor ENST00000624431.2
## [3] chr1 183215-183222 + | donor ENST00000624431.2
## [4] chr1 183490-183494 + | acceptor ENST00000624431.2
## [5] chr1 183570-183577 + | donor ENST00000624431.2
## ... ... ... ... . ... ...
## [726] chr1 810061-810068 - | donor ENST00000590817.3
## [727] chr1 810170-810174 - | acceptor ENST00000447500.4
## [728] chr1 817367-817374 - | donor ENST00000447500.4
## [729] chr1 827664-827671 - | donor ENST00000635509.2
## [730] chr1 827664-827671 - | donor ENST00000634337.2
## transcript_name gene_id gene_name exon_number
## <character> <character> <character> <integer>
## [1] DDX11L17-201 ENSG00000279928.2 DDX11L17 1
## [2] DDX11L17-201 ENSG00000279928.2 DDX11L17 2
## [3] DDX11L17-201 ENSG00000279928.2 DDX11L17 2
## [4] DDX11L17-201 ENSG00000279928.2 DDX11L17 3
## [5] DDX11L17-201 ENSG00000279928.2 DDX11L17 3
## ... ... ... ... ...
## [726] ENST00000590817 ENSG00000230092.8 ENSG00000230092 1
## [727] ENST00000447500 ENSG00000290784.1 ENSG00000290784 2
## [728] ENST00000447500 ENSG00000290784.1 ENSG00000290784 1
## [729] ENST00000635509 ENSG00000230021.10 ENSG00000230021 1
## [730] ENST00000634337 ENSG00000230021.10 ENSG00000230021 1
## -------
## seqinfo: 1 sequence from GRCh38 genomeMany splice junctions will be shared across multiple transcripts, so
the returned set of junctions can also be simplified using
chopMC() from extraChIPs.
chopMC(sj)## GRanges object with 196 ranges and 6 metadata columns:
## seqnames ranges strand | site
## <Rle> <IRanges> <Rle> | <character>
## [1] chr1 182745-182752 + | donor
## [2] chr1 183128-183132 + | acceptor
## [3] chr1 183215-183222 + | donor
## [4] chr1 183490-183494 + | acceptor
## [5] chr1 183570-183577 + | donor
## ... ... ... ... . ...
## [192] chr1 808623-808627 - | acceptor
## [193] chr1 810061-810068 - | donor
## [194] chr1 810170-810174 - | acceptor
## [195] chr1 817367-817374 - | donor
## [196] chr1 827664-827671 - | donor
## transcript_id transcript_name
## <CharacterList> <CharacterList>
## [1] ENST00000624431.2 DDX11L17-201
## [2] ENST00000624431.2 DDX11L17-201
## [3] ENST00000624431.2 DDX11L17-201
## [4] ENST00000624431.2 DDX11L17-201
## [5] ENST00000624431.2 DDX11L17-201
## ... ... ...
## [192] ENST00000447500.4,ENST00000590817.3 ENST00000447500,ENST00000590817
## [193] ENST00000447500.4,ENST00000590817.3 ENST00000447500,ENST00000590817
## [194] ENST00000447500.4 ENST00000447500
## [195] ENST00000447500.4 ENST00000447500
## [196] ENST00000635509.2,ENST00000634337.2 ENST00000635509,ENST00000634337
## gene_id gene_name
## <CharacterList> <CharacterList>
## [1] ENSG00000279928.2 DDX11L17
## [2] ENSG00000279928.2 DDX11L17
## [3] ENSG00000279928.2 DDX11L17
## [4] ENSG00000279928.2 DDX11L17
## [5] ENSG00000279928.2 DDX11L17
## ... ... ...
## [192] ENSG00000290784.1,ENSG00000230092.8 ENSG00000290784,ENSG00000230092
## [193] ENSG00000290784.1,ENSG00000230092.8 ENSG00000290784,ENSG00000230092
## [194] ENSG00000290784.1 ENSG00000290784
## [195] ENSG00000290784.1 ENSG00000290784
## [196] ENSG00000230021.10,ENSG00000230021.10 ENSG00000230021,ENSG00000230021
## exon_number
## <IntegerList>
## [1] 1
## [2] 2
## [3] 2
## [4] 3
## [5] 3
## ... ...
## [192] 3,2
## [193] 2,1
## [194] 2
## [195] 1
## [196] 1,1
## -------
## seqinfo: 1 sequence from an unspecified genome; no seqlengthsAs a further alternative, splice junctions can be returned as a set
of interactions, with donor sites being assigned to the
anchorOne element, and acceptor sites being placed in the
anchorTwo element This enables the identification of all
splice junctions for specific transcripts.
sj <- sjFromExons(gtf$exon, extra_cols = ec, as = "GInteractions")
subset(sj, transcript_name == "DDX11L17-201")## GInteractions object with 4 interactions and 5 metadata columns:
## seqnames1 ranges1 seqnames2 ranges2 | transcript_id
## <Rle> <IRanges> <Rle> <IRanges> | <character>
## [1] chr1 182745-182752 --- chr1 183128-183132 | ENST00000624431.2
## [2] chr1 183215-183222 --- chr1 183490-183494 | ENST00000624431.2
## [3] chr1 183570-183577 --- chr1 183736-183740 | ENST00000624431.2
## [4] chr1 183900-183907 --- chr1 183977-183981 | ENST00000624431.2
## transcript_name gene_id gene_name sj
## <character> <character> <character> <integer>
## [1] DDX11L17-201 ENSG00000279928.2 DDX11L17 1
## [2] DDX11L17-201 ENSG00000279928.2 DDX11L17 2
## [3] DDX11L17-201 ENSG00000279928.2 DDX11L17 3
## [4] DDX11L17-201 ENSG00000279928.2 DDX11L17 4
## -------
## regions: 196 ranges and 0 metadata columns
## seqinfo: 1 sequence from GRCh38 genomeSession info
## R version 4.5.1 (2025-06-13)
## Platform: x86_64-pc-linux-gnu
## Running under: Ubuntu 24.04.3 LTS
##
## Matrix products: default
## BLAS: /usr/lib/x86_64-linux-gnu/openblas-pthread/libblas.so.3
## LAPACK: /usr/lib/x86_64-linux-gnu/openblas-pthread/libopenblasp-r0.3.26.so; LAPACK version 3.12.0
##
## locale:
## [1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
## [3] LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8
## [5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
## [7] LC_PAPER=en_US.UTF-8 LC_NAME=C
## [9] LC_ADDRESS=C LC_TELEPHONE=C
## [11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
##
## time zone: UTC
## tzcode source: system (glibc)
##
## attached base packages:
## [1] stats4 stats graphics grDevices utils datasets methods
## [8] base
##
## other attached packages:
## [1] BSgenome.Hsapiens.UCSC.hg38_1.4.5 BSgenome_1.77.2
## [3] BiocIO_1.19.0 GenomeInfoDb_1.45.12
## [5] GenomicFeatures_1.61.6 AnnotationDbi_1.71.2
## [7] transmogR_1.5.2 extraChIPs_1.13.3
## [9] tibble_3.3.0 ggside_0.4.0
## [11] ggplot2_4.0.0 BiocParallel_1.43.4
## [13] rtracklayer_1.69.1 VariantAnnotation_1.55.2
## [15] Rsamtools_2.25.3 Biostrings_2.77.2
## [17] XVector_0.49.1 SummarizedExperiment_1.39.2
## [19] Biobase_2.69.1 GenomicRanges_1.61.5
## [21] IRanges_2.43.5 S4Vectors_0.47.4
## [23] Seqinfo_0.99.2 MatrixGenerics_1.21.0
## [25] matrixStats_1.5.0 BiocGenerics_0.55.4
## [27] generics_0.1.4 BiocStyle_2.37.1
##
## loaded via a namespace (and not attached):
## [1] DBI_1.2.3 bitops_1.0-9 SimpleUpset_0.1.2
## [4] rlang_1.1.6 magrittr_2.0.4 compiler_4.5.1
## [7] RSQLite_2.4.3 png_0.1-8 systemfonts_1.3.1
## [10] vctrs_0.6.5 stringr_1.5.2 pkgconfig_2.0.3
## [13] crayon_1.5.3 fastmap_1.2.0 labeling_0.4.3
## [16] rmarkdown_2.30 UCSC.utils_1.5.0 ragg_1.5.0
## [19] purrr_1.1.0 bit_4.6.0 xfun_0.53
## [22] cachem_1.1.0 jsonlite_2.0.0 blob_1.2.4
## [25] DelayedArray_0.35.3 parallel_4.5.1 R6_2.6.1
## [28] bslib_0.9.0 stringi_1.8.7 RColorBrewer_1.1-3
## [31] limma_3.65.6 jquerylib_0.1.4 Rcpp_1.1.0
## [34] bookdown_0.45 knitr_1.50 Matrix_1.7-4
## [37] tidyselect_1.2.1 abind_1.4-8 yaml_2.3.10
## [40] codetools_0.2-20 curl_7.0.0 lattice_0.22-7
## [43] InteractionSet_1.37.1 withr_3.0.2 KEGGREST_1.49.2
## [46] S7_0.2.0 csaw_1.43.1 evaluate_1.0.5
## [49] desc_1.4.3 pillar_1.11.1 BiocManager_1.30.26
## [52] RCurl_1.98-1.17 scales_1.4.0 glue_1.8.0
## [55] metapod_1.17.0 tools_4.5.1 data.table_1.17.8
## [58] locfit_1.5-9.12 GenomicAlignments_1.45.4 forcats_1.0.1
## [61] fs_1.6.6 XML_3.99-0.19 grid_4.5.1
## [64] tidyr_1.3.1 edgeR_4.7.6 patchwork_1.3.2
## [67] restfulr_0.0.16 cli_3.6.5 textshaping_1.0.4
## [70] S4Arrays_1.9.1 dplyr_1.1.4 gtable_0.3.6
## [73] sass_0.4.10 digest_0.6.37 SparseArray_1.9.1
## [76] ggrepel_0.9.6 rjson_0.2.23 htmlwidgets_1.6.4
## [79] farver_2.1.2 memoise_2.0.1 htmltools_0.5.8.1
## [82] pkgdown_2.1.3.9000 lifecycle_1.0.4 httr_1.4.7
## [85] statmod_1.5.1 bit64_4.6.0-1