QC: Trimmed Data
12 July, 2023
library(tidyverse)
library(glue)
library(yaml)
library(here)
library(reactable)
library(pander)
library(ngsReports)
library(scales)
library(htmltools)
library(Polychrome)
myTheme <- theme(
plot.title = element_text(hjust = 0.5),
text = element_text(size = 13)
)config <- read_yaml(here::here("config/config.yml"))
samples <- read_tsv(here::here(config$samples)) %>%
bind_rows(
tibble(
accession = unique(.$input), target = "Input"
)
)
n <- length(samples$accession)
pal <- createPalette(n, c("#2A95E8", "#E5629C"), range = c(10, 60), M = 100000)
names(pal) <- samples$accession
colours <- scale_colour_manual(values = pal)
qc_path <- here::here(config$paths$qc, "trimmed")
rel_qc_path <- ifelse(
grepl("docs", qc_path), gsub(".+docs", ".", qc_path),
gsub(here::here(), "..", qc_path)
)
fl <- file.path(qc_path, glue("{samples$accession}_fastqc.zip")) %>%
setNames(samples$accession)
trimFqc <- FastqcDataList(fl[file.exists(fl)])
names(trimFqc) <- names(fl)[file.exists(fl)]Introduction
This page provides summary statistics and diagnostics for the data after trimming with AdapterRemoval (Schubert, Lindgreen, and Orlando 2016) Most data was obtained from FastQC and parsed using the Bioconductor package ngsReports (Ward, To, and Pederson 2020). FastQC reports for 19 files were found.
A conventional MultiQC report (Ewels et al. 2016) can also be found here
Data Summary
div(
class = "table",
div(
class = "table-header",
htmltools::tags$caption(
htmltools::em(
"Library sizes with links to all FastQC reports generated after
processing with AdapterRemoval"
)
)
),
readTotals(trimFqc) %>%
mutate(Filename = str_remove_all(Filename, ".(fast|f)q.gz")) %>%
left_join(samples, by = c("Filename" = "accession")) %>%
dplyr::select(-input) %>%
setNames(str_replace_all(names(.), "_", " ")) %>%
setNames(str_to_title(names(.))) %>%
reactable(
sortable = TRUE, resizable = TRUE,
showPageSizeOptions = TRUE,
columns = list(
Filename = colDef(
cell = function(value) htmltools::tags$a(
href = file.path(rel_qc_path, glue("{value}_fastqc.html")),
target = "_blank",
value
),
html = TRUE
)
),
defaultColDef = colDef(format = colFormat(separators = TRUE))
)
)FastQC Status
plotSummary(trimFqc) + myTheme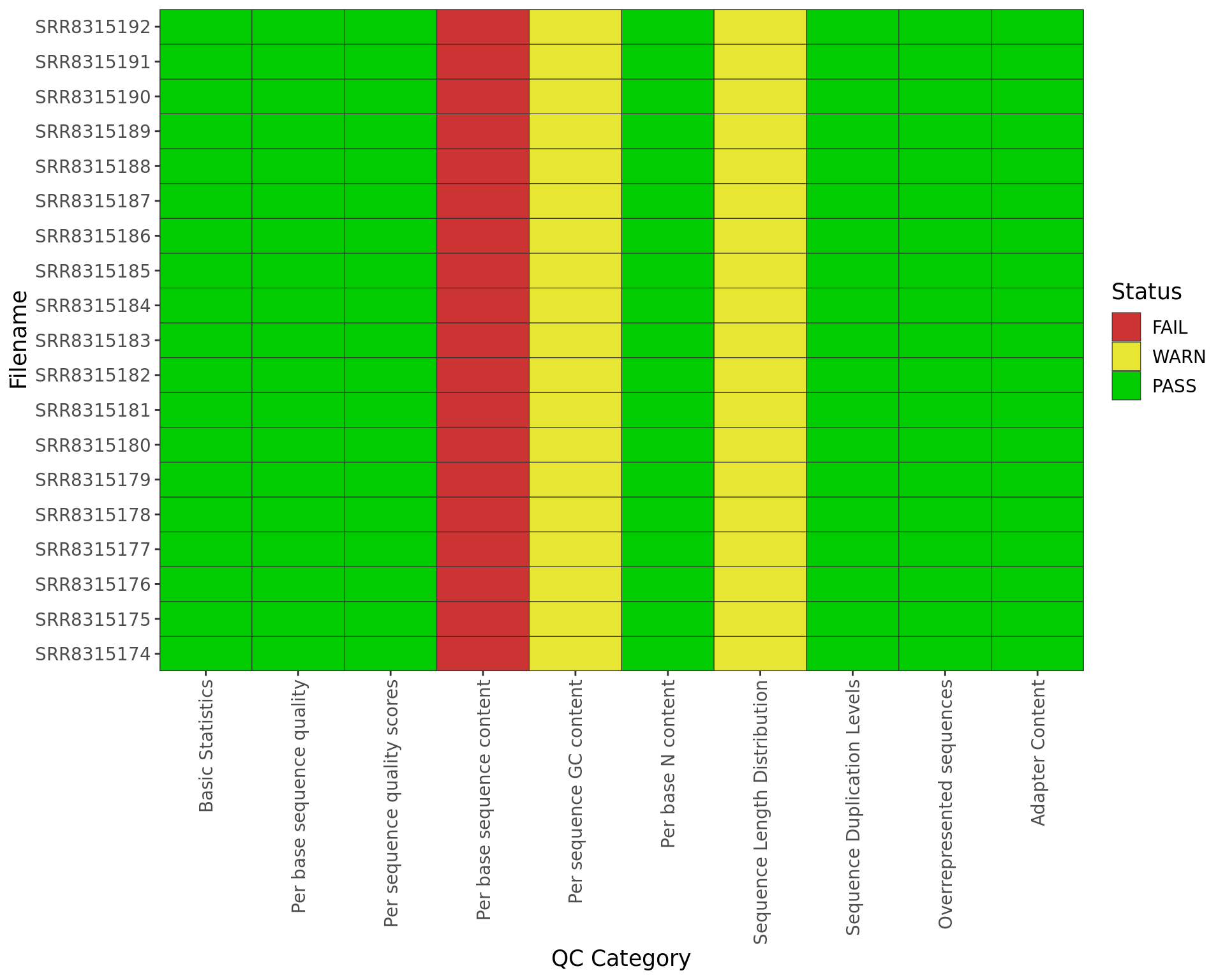
Summary of Pass/Warn/Fail status from all samples after trimming
Trimming Statistics
trim_logs <- here::here(
"output", "adapterremoval", glue("{samples$accession}.settings")
) %>%
setNames(samples$accession) %>%
.[file.exists(.)] %>%
importNgsLogs(which = "statistics") %>%
mutate(Filename = str_remove_all(Filename, ".settings")) %>%
left_join(samples, by = c("Filename" = "accession"))
maxAdapt <- maxAdapterContent(trimFqc, asPercent = FALSE) %>%
ungroup() %>%
dplyr::select(-Filename) %>%
lapply(range) %>%
.[vapply(., max, numeric(1)) > 0] %>%
lapply(percent, accuracy = 0.01)Retained Reads
max_reads <- max(trim_logs$`Total number of reads`)
trim_logs %>%
dplyr::select(
Filename, target, treatment,
Retained = `Number of retained reads`,
Discarded = `Number of discarded mate 1 reads`
) %>%
pivot_longer(cols = ends_with("ed"), names_to = "Status", values_to = "Reads") %>%
mutate(Status = as.factor(Status)) %>%
arrange(Filename, desc(Status)) %>%
mutate(
y = cumsum(Reads), p = Reads / sum(Reads), .by = Filename,
treatment = str_replace_na(treatment, "")
) %>%
mutate(y = ifelse(Status == "Retained", 0.925 * y, y - 0.5*Reads)) %>%
ggplot(aes(Filename, Reads, fill = Status)) +
geom_col() +
geom_label(
aes(Filename, y, label = percent(p, 0.1)),
size = 3.5, alpha = 0.7, fill = "white", show.legend = FALSE
) +
facet_grid(target +treatment ~ ., scales = "free", space = "free") +
scale_y_continuous(
labels = comma_format(scale = 1e-6), expand = expansion(c(0, 0.05))
) +
scale_fill_brewer(palette = "Set1") +
labs(y = "Reads (millions)") +
theme_bw() +
myTheme +
coord_flip()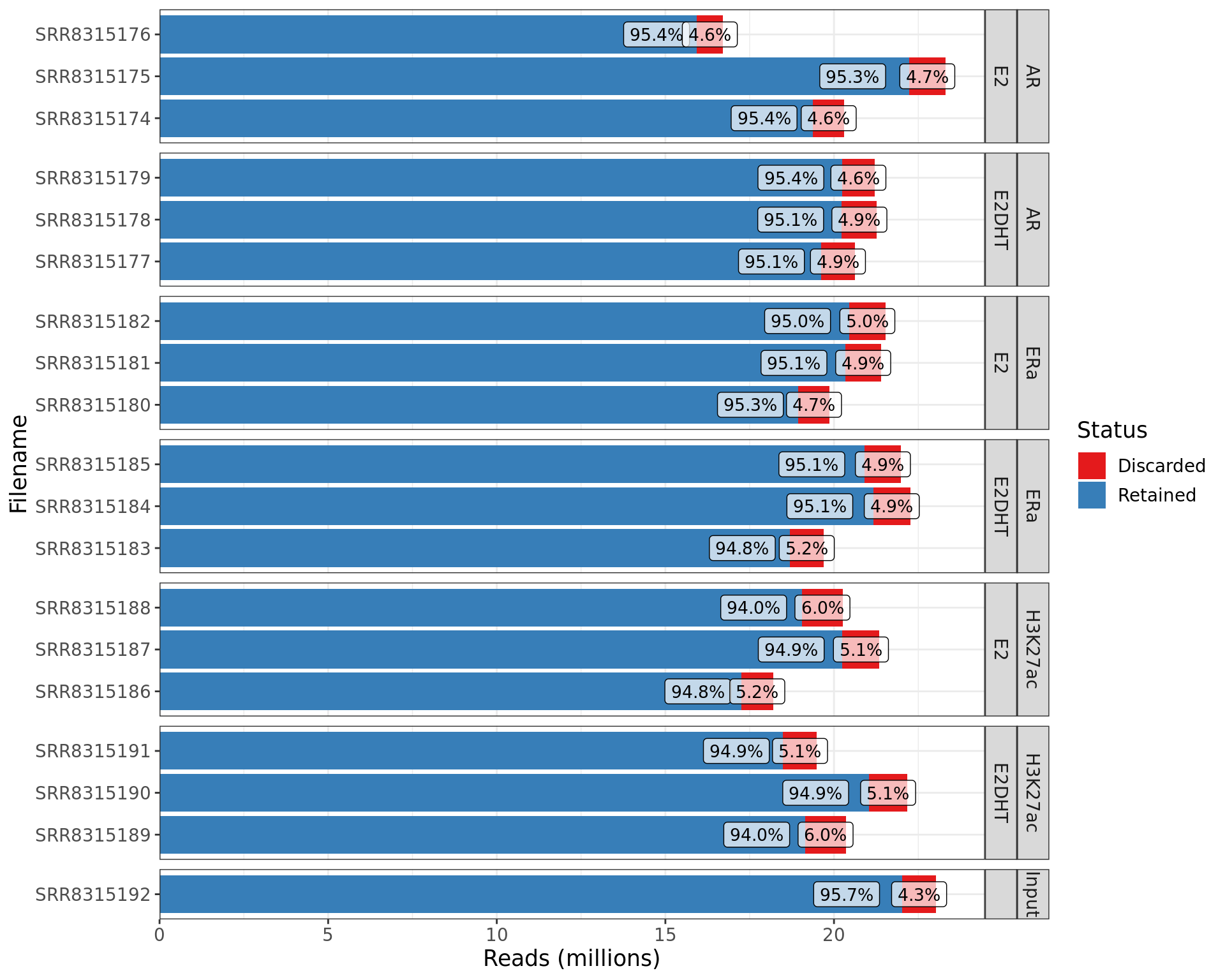
Summary of retained and discarded reads from each library. The percentage of reads retained or discarded in each library is shown using labels.
Of the entire set of files, the ranges of values for all possible remaining adapter sequences were
- **Illumina_Small_RNA_3’_Adapter**: 0.00% and 0.02%
- **Illumina_Small_RNA_5’_Adapter**: 0.00% and 0.01%
- Nextera_Transposase_Sequence: 0.06% and 0.09%
- SOLID_Small_RNA_Adapter: 0.00% and 0.04%
noting that some of these putative adapters may be completely artefactual and only sequences matching the provided adapter sequence were removed.
QC Diagnostics
Quality Scores
plotBaseQuals(trimFqc, heat_w = 20, dendrogram = TRUE, usePlotly = TRUE, text = element_text(size = 13))Mean Quality Scores are each position along the reads
Sequence Content Heatmap
plotSeqContent(trimFqc, usePlotly = TRUE, dendrogram = TRUE, heat_w = 20, text = element_text(size = 13))Sequence content along each read
Sequence Content Residuals
All Libraries
plotSeqContent(trimFqc, plotType = "residuals", scaleColour = colours) + myTheme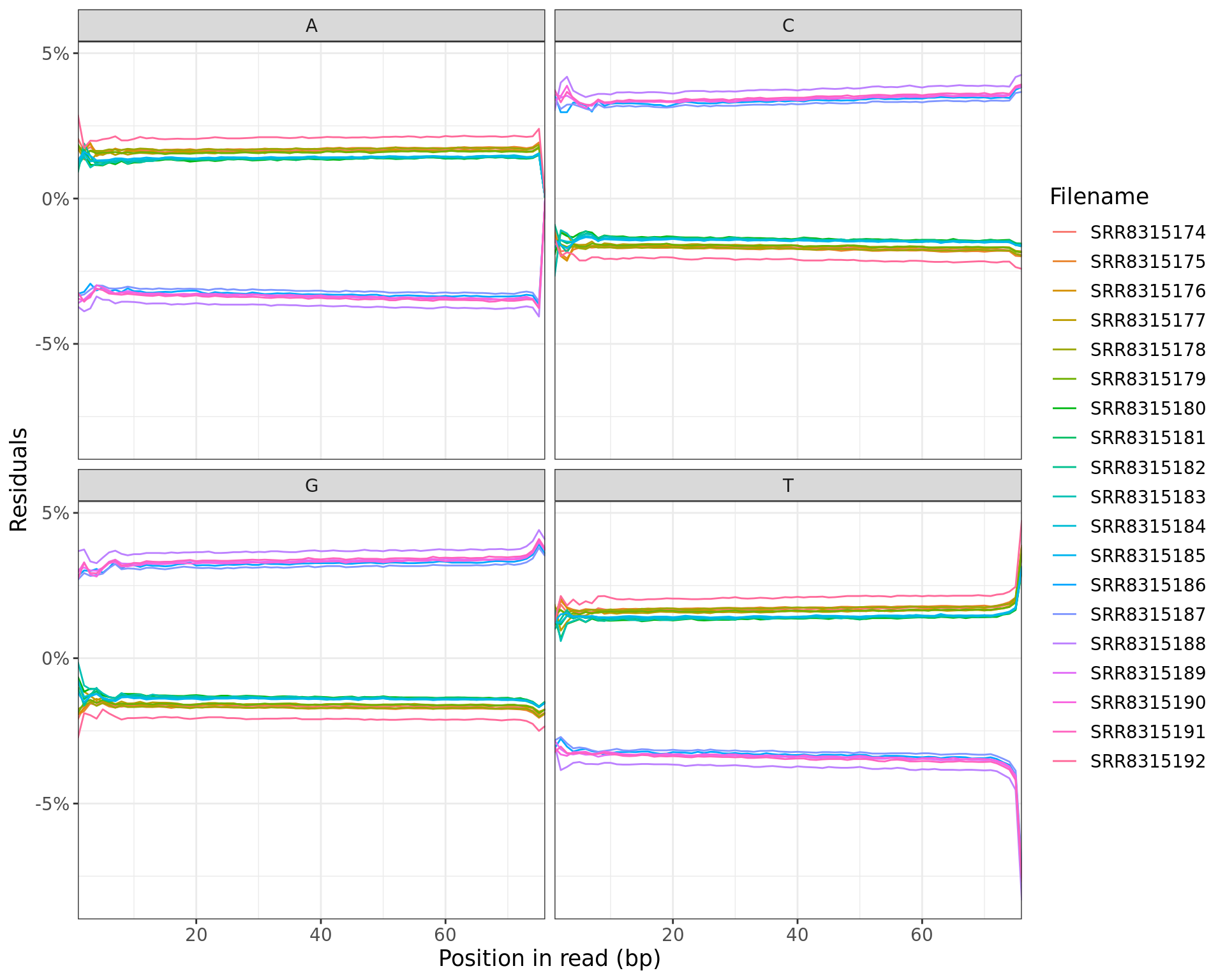
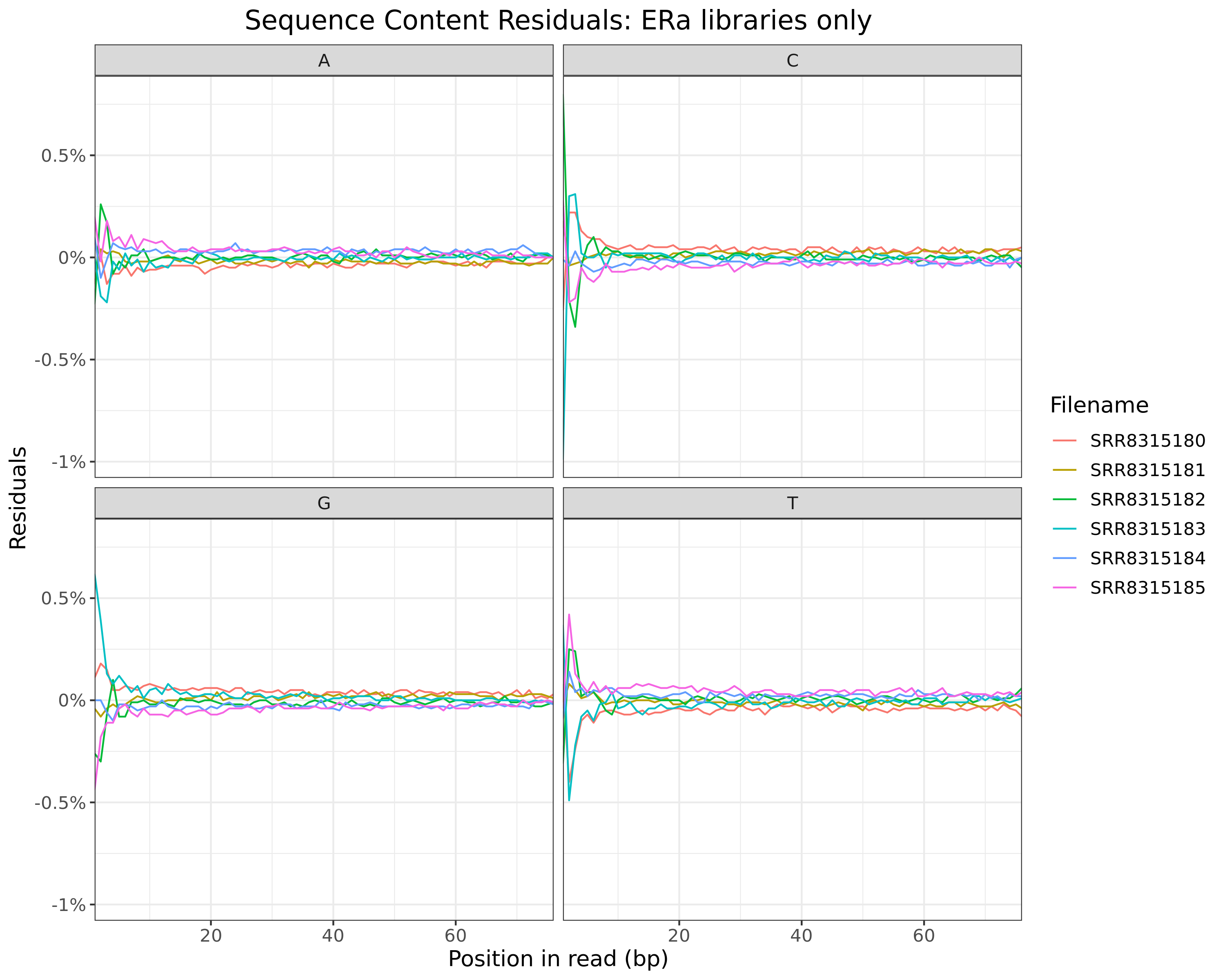
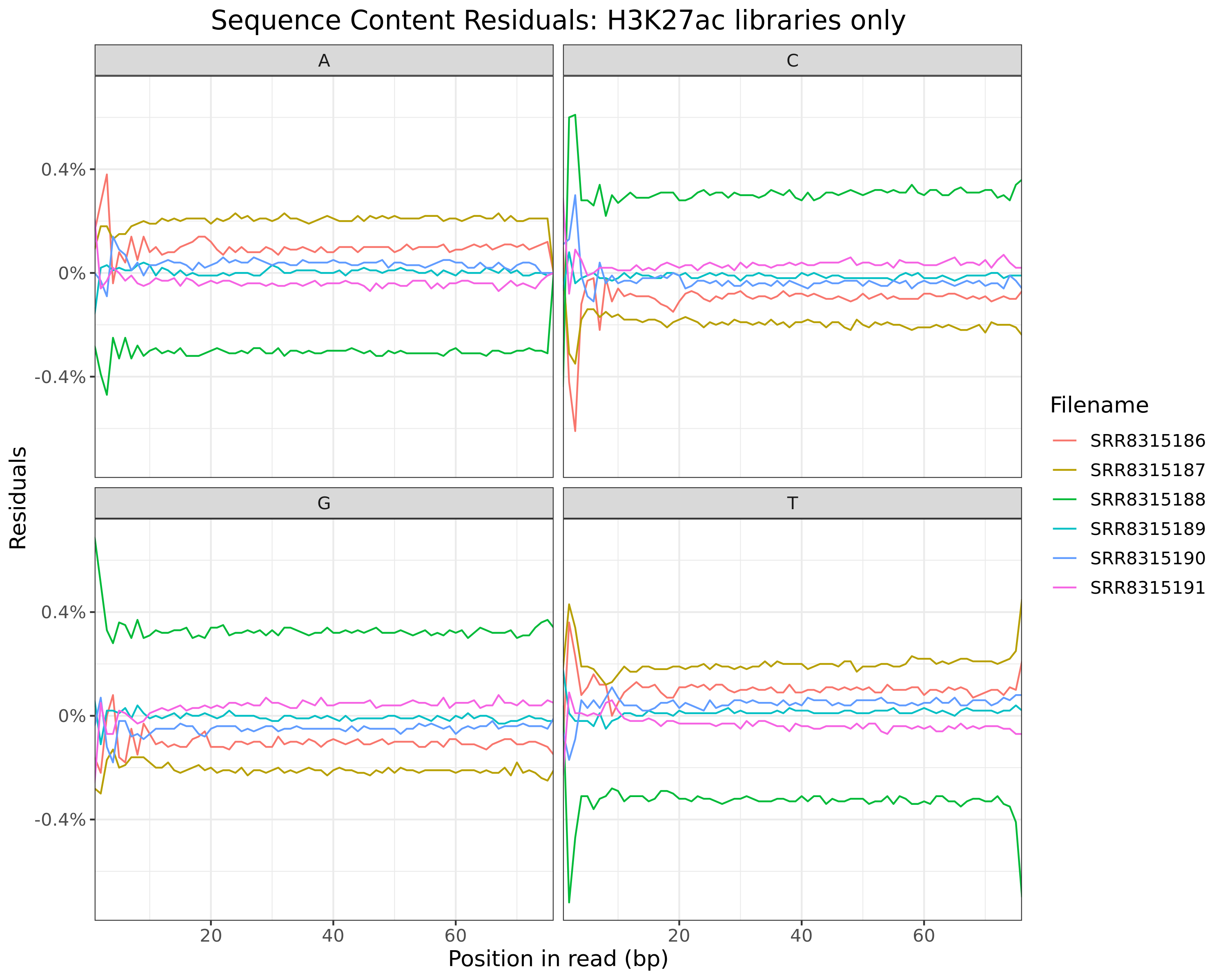
Residuals obtained when subtracting mean values at each position
d <- here::here("docs", "assets", "trimmed_fqc")
if (!dir.exists(d)) dir.create(d, recursive = TRUE)
h <- knitr::opts_chunk$get("fig.height")
w <- knitr::opts_chunk$get("fig.width")
htmltools::tagList(
samples %>%
dplyr::filter(accession %in% names(trimFqc)) %>%
split(.$target) %>%
setNames(NULL) %>%
lapply(
function(x) {
tgt <- unique(x$target)
fqc <- trimFqc[x$accession]
p <- plotSeqContent(fqc, plotType = "residuals", scaleColour = colours) +
ggtitle(glue("Sequence Content Residuals: {tgt} libraries only")) +
myTheme
png_out <- file.path(
d, glue("seq-content-residuals-{tgt}.png")
)
href <- str_extract(png_out, "assets.+")
png(filename = png_out, width = w, height = h, units = "in", res = 300)
print(p)
dev.off()
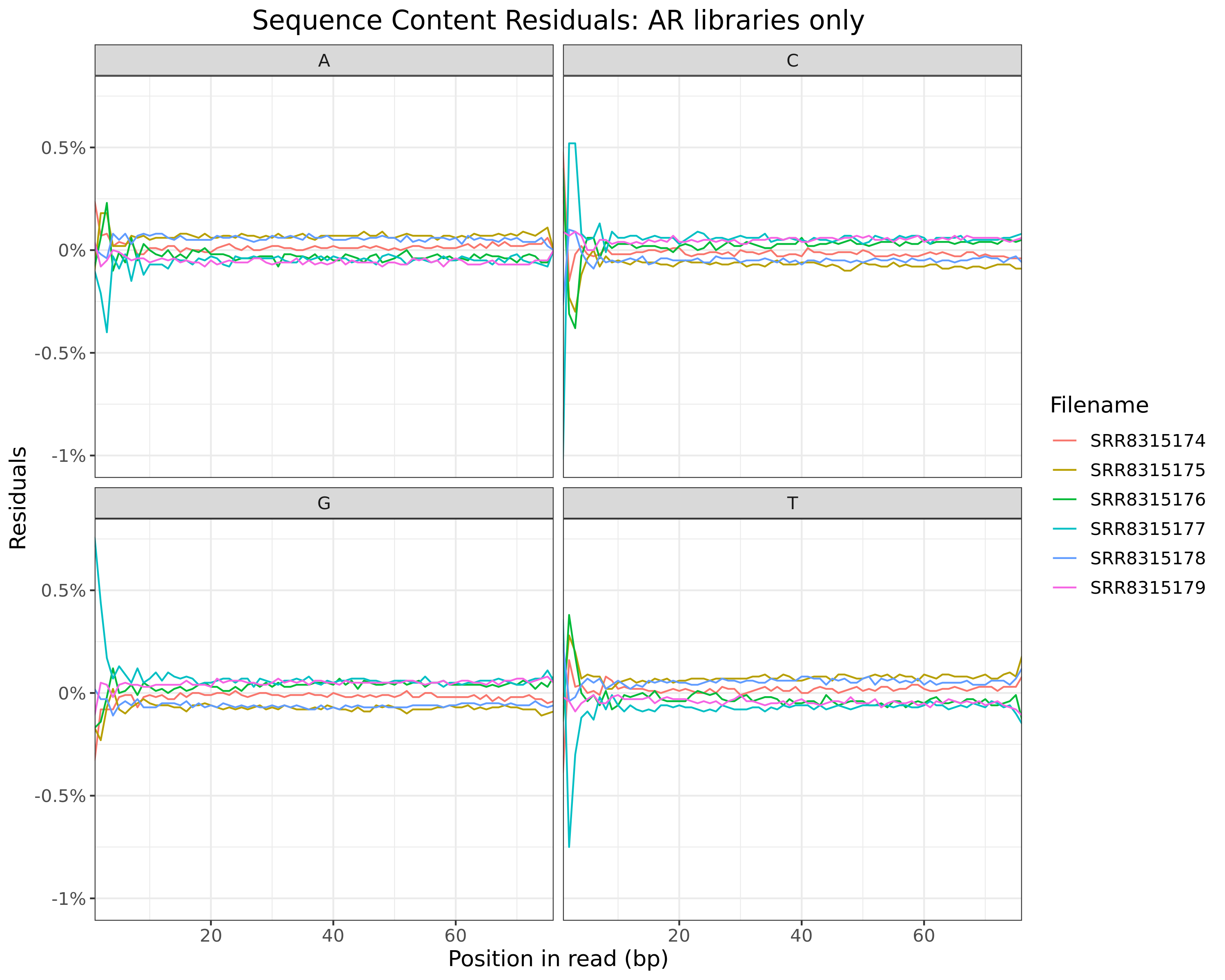
cp <- htmltools::em(
glue(
"
Residuals obtained when subtracting mean values at each position for
{tgt} libraries only.
"
)
)
htmltools::div(
htmltools::div(
id = glue("plot-seq-residuals-{tgt}"),
class = "section level4",
htmltools::h4(tgt),
htmltools::div(
class = "figure", style = "text-align: center",
htmltools::img(src = href, width = "100%"),
htmltools::tags$caption(cp)
)
)
)
}
)
)AR
ERa
H3K27ac
Input
GC Content
All Libraries
plotGcContent(
trimFqc, usePlotly = TRUE, plotType = "line", theoreticalGC = FALSE,
scaleColour = colours, plotlyLegend = TRUE, text = element_text(size = 13)
)GC content distributions for all libraries
h <- knitr::opts_chunk$get("fig.height")
w <- knitr::opts_chunk$get("fig.width")
htmltools::tagList(
samples %>%
dplyr::filter(accession %in% names(trimFqc)) %>%
split(.$target) %>%
setNames(NULL) %>%
lapply(
function(x) {
tgt <- unique(x$target)
fqc <- trimFqc[x$accession]
p <- plotGcContent(
fqc, plotType = "line", theoreticalGC = FALSE, scaleColour = colours
) +
ggtitle(glue("GC Content Distribuions: {tgt} libraries only")) +
myTheme
png_out <- file.path(
d, glue("gc-content-{tgt}.png")
)
href <- str_extract(png_out, "assets.+")
png(filename = png_out, width = w, height = h, units = "in", res = 300)
print(p)
dev.off()
cp <- htmltools::em(
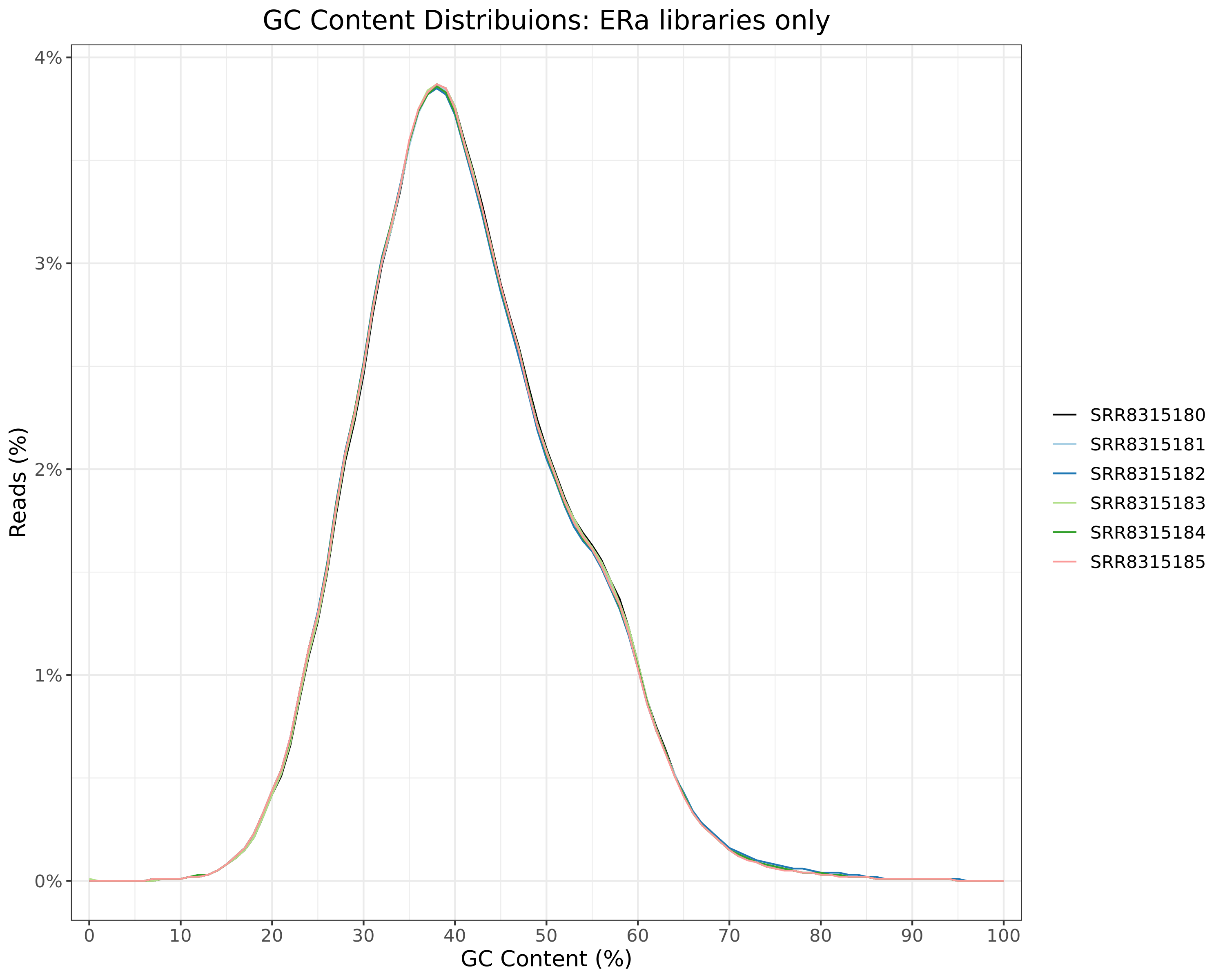
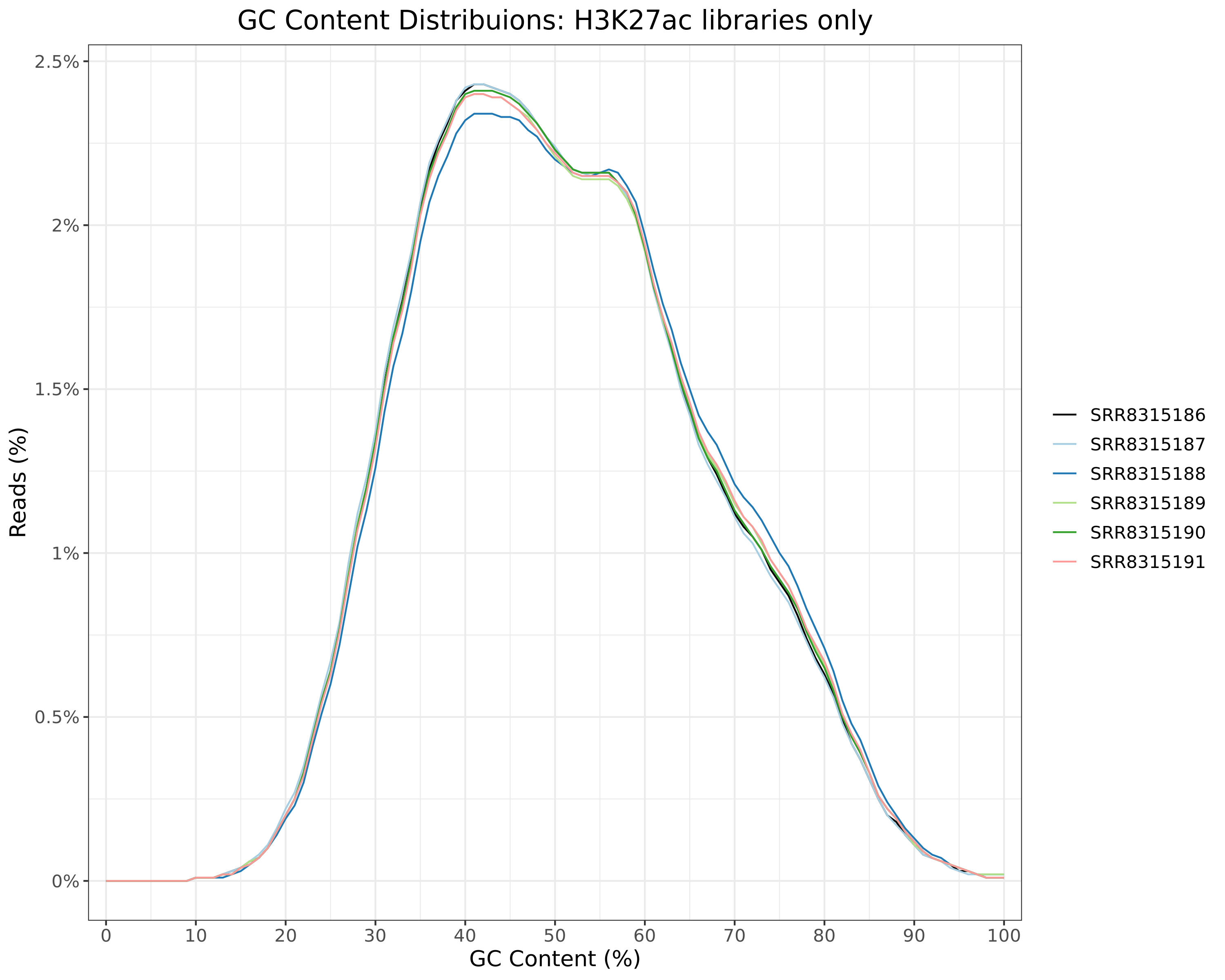
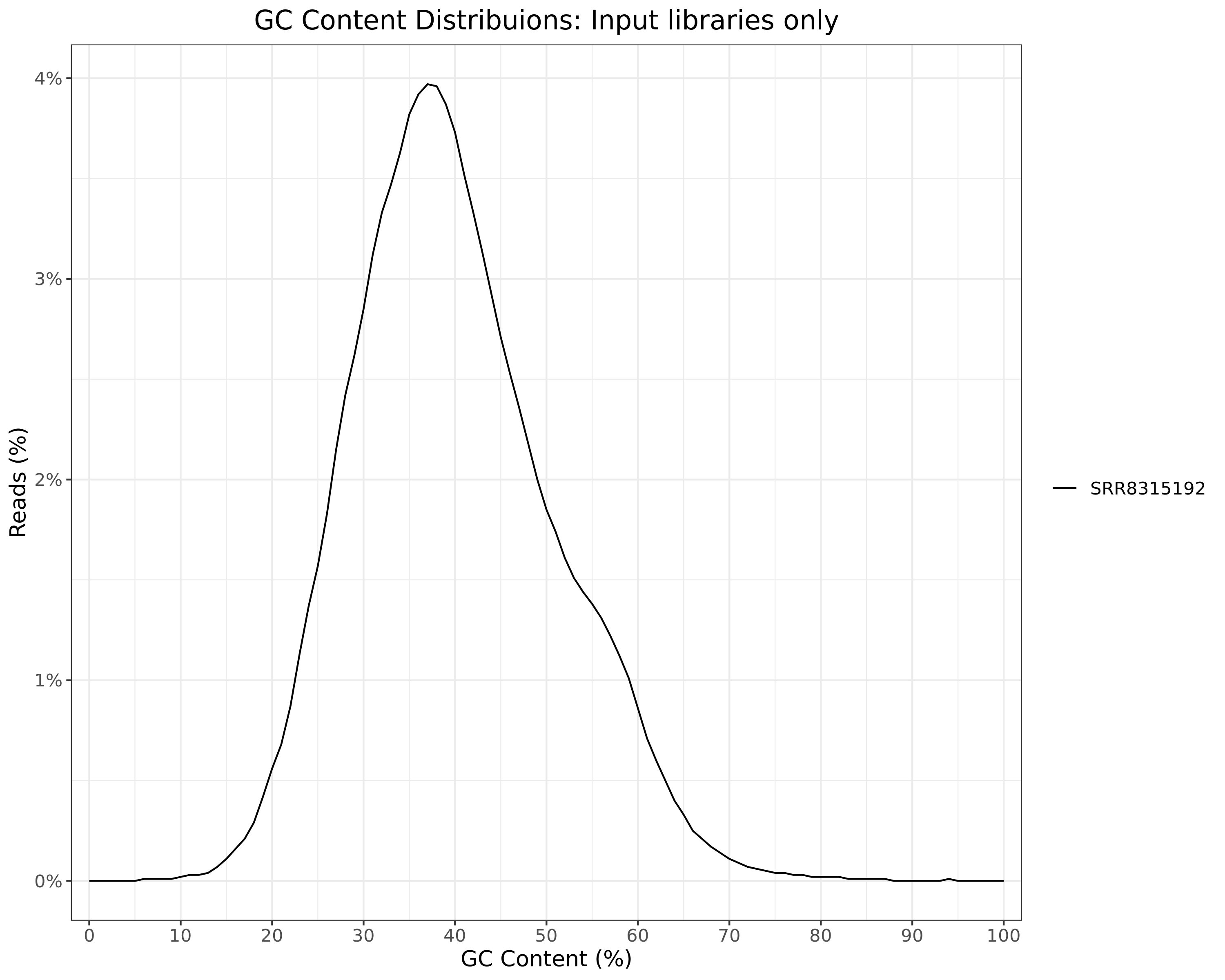
glue("GC content distributions for {tgt} libraries only.")
)
htmltools::div(
htmltools::div(
id = glue("plot-gc_content-{tgt}"),
class = "section level4",
htmltools::h4(tgt),
htmltools::div(
class = "figure", style = "text-align: center",
htmltools::img(src = href, width = "100%"),
htmltools::tags$caption(cp)
)
)
)
}
)
)AR
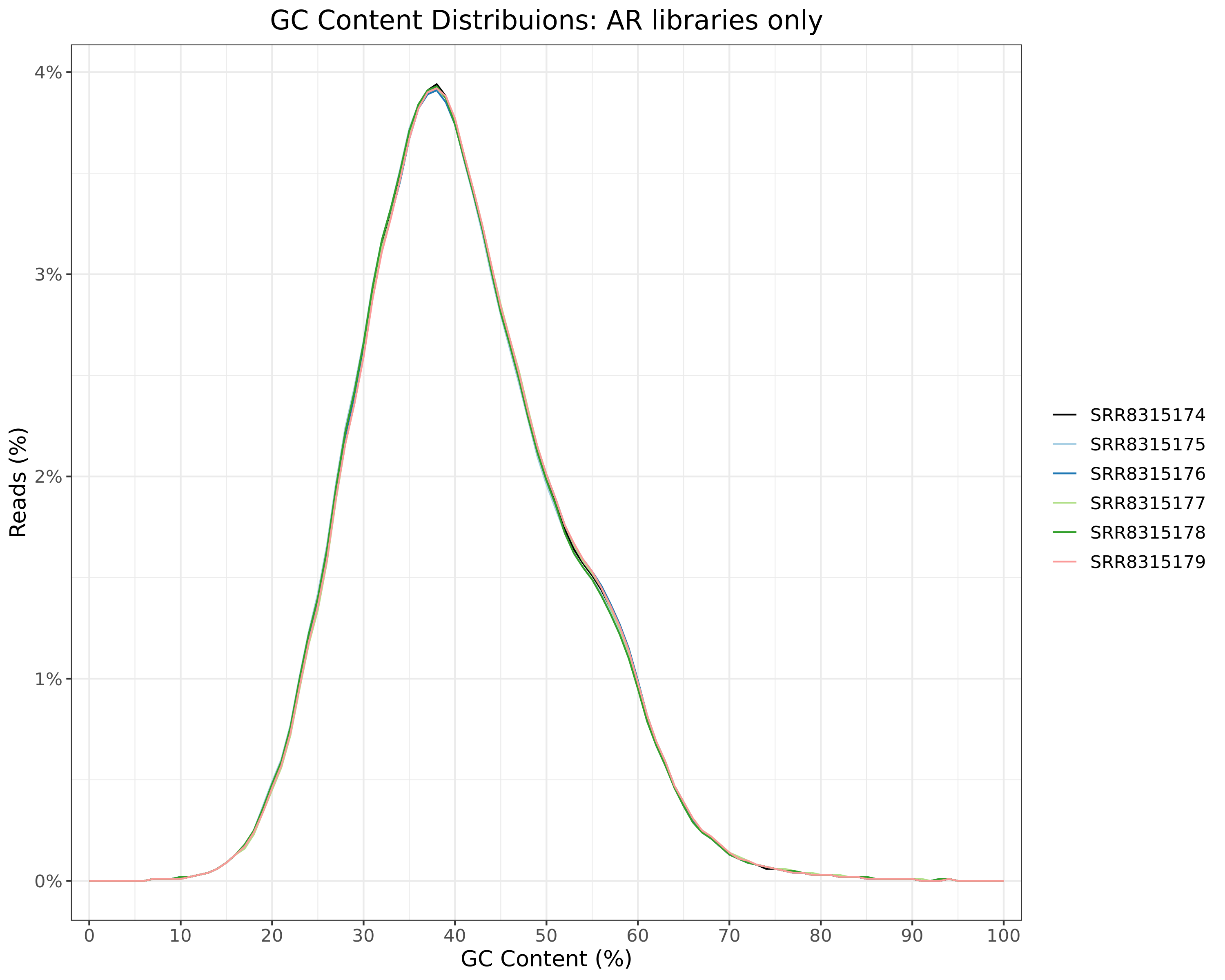
ERa
H3K27ac
Input
Sequence Lengths
plotSeqLengthDistn(
trimFqc, usePlotly = TRUE, plotType = "cdf", scaleColour = colours,
plotlyLegend = TRUE, text = element_text(size = 13)
)Cumulative Sequence Length distributions
Duplication Levels
plotDupLevels(
trimFqc, usePlotly = TRUE, dendrogram = TRUE, heat_w = 20,
text = element_text(size = 13)
)Duplication levels as estimated by FastQC
References
R version 4.2.3 (2023-03-15)
Platform: x86_64-conda-linux-gnu (64-bit)
locale: LC_CTYPE=en_AU.UTF-8, LC_NUMERIC=C, LC_TIME=en_AU.UTF-8, LC_COLLATE=en_AU.UTF-8, LC_MONETARY=en_AU.UTF-8, LC_MESSAGES=en_AU.UTF-8, LC_PAPER=en_AU.UTF-8, LC_NAME=C, LC_ADDRESS=C, LC_TELEPHONE=C, LC_MEASUREMENT=en_AU.UTF-8 and LC_IDENTIFICATION=C
attached base packages: stats, graphics, grDevices, utils, datasets, methods and base
other attached packages: Polychrome(v.1.5.1), htmltools(v.0.5.5), scales(v.1.2.1), ngsReports(v.2.0.0), patchwork(v.1.1.2), BiocGenerics(v.0.44.0), pander(v.0.6.5), reactable(v.0.4.4), here(v.1.0.1), yaml(v.2.3.7), glue(v.1.6.2), lubridate(v.1.9.2), forcats(v.1.0.0), stringr(v.1.5.0), dplyr(v.1.1.2), purrr(v.1.0.1), readr(v.2.1.4), tidyr(v.1.3.0), tibble(v.3.2.1), ggplot2(v.3.4.2) and tidyverse(v.2.0.0)
loaded via a namespace (and not attached): ggdendro(v.0.1.23), httr(v.1.4.6), sass(v.0.4.6), bit64(v.4.0.5), vroom(v.1.6.3), jsonlite(v.1.8.5), viridisLite(v.0.4.2), bslib(v.0.5.0), highr(v.0.10), stats4(v.4.2.3), GenomeInfoDbData(v.1.2.9), pillar(v.1.9.0), lattice(v.0.21-8), digest(v.0.6.31), RColorBrewer(v.1.1-3), XVector(v.0.38.0), colorspace(v.2.1-0), plyr(v.1.8.8), reactR(v.0.4.4), pkgconfig(v.2.0.3), zlibbioc(v.1.44.0), tzdb(v.0.4.0), timechange(v.0.2.0), farver(v.2.1.1), generics(v.0.1.3), IRanges(v.2.32.0), ellipsis(v.0.3.2), DT(v.0.28), cachem(v.1.0.8), withr(v.2.5.0), lazyeval(v.0.2.2), cli(v.3.6.1), magrittr(v.2.0.3), crayon(v.1.5.2), evaluate(v.0.21), fansi(v.1.0.4), MASS(v.7.3-60), tools(v.4.2.3), data.table(v.1.14.8), hms(v.1.1.3), lifecycle(v.1.0.3), plotly(v.4.10.2), S4Vectors(v.0.36.0), munsell(v.0.5.0), Biostrings(v.2.66.0), compiler(v.4.2.3), jquerylib(v.0.1.4), GenomeInfoDb(v.1.34.9), rlang(v.1.1.1), grid(v.4.2.3), RCurl(v.1.98-1.12), rstudioapi(v.0.14), htmlwidgets(v.1.6.2), crosstalk(v.1.2.0), labeling(v.0.4.2), bitops(v.1.0-7), rmarkdown(v.2.21), gtable(v.0.3.3), reshape2(v.1.4.4), R6(v.2.5.1), zoo(v.1.8-12), knitr(v.1.43), fastmap(v.1.1.1), bit(v.4.0.5), utf8(v.1.2.3), rprojroot(v.2.0.3), stringi(v.1.7.12), parallel(v.4.2.3), Rcpp(v.1.0.10), vctrs(v.0.6.3), scatterplot3d(v.0.3-44), tidyselect(v.1.2.0) and xfun(v.0.39)