ER: MACS2 Summary
06 July, 2023
target <- params$target
threads <- params$threadslibrary(tidyverse)
library(magrittr)
library(rtracklayer)
library(glue)
library(pander)
library(scales)
library(plyranges)
library(yaml)
library(ngsReports)
library(ComplexUpset)
library(VennDiagram)
library(rlang)
library(BiocParallel)
library(parallel)
library(Rsamtools)
library(Biostrings)
library(ggside)
library(extraChIPs)
library(patchwork)
panderOptions("big.mark", ",")
panderOptions("missing", "")
panderOptions("table.split.table", Inf)
theme_set(
theme_bw() +
theme(
plot.title = element_text(hjust = 0.5)
)
)
register(MulticoreParam(workers = threads))
source(here::here("workflow/scripts/custom_functions.R"))config <- read_yaml(
here::here("config", "config.yml")
)
fdr_alpha <- config$comparisons$fdr
extra_params <- read_yaml(here::here("config", "params.yml"))
bam_path <- here::here(config$paths$bam)
macs2_path <- here::here("output", "macs2")
annotation_path <- here::here("output", "annotations")
colours <- read_rds(
file.path(annotation_path, "colours.rds")
) %>%
lapply(unlist)samples <- macs2_path %>%
file.path(target, glue("{target}_qc_samples.tsv")) %>%
read_tsv()
treat_levels <- unique(samples$treat)
if (!is.null(config$comparisons$contrasts)) {
## Ensure levels respect those provided in contrasts
treat_levels <- config$comparisons$contrasts %>%
unlist() %>%
intersect(samples$treat) %>%
unique()
}
rep_col <- setdiff(
colnames(samples), c("sample", "treat", "target", "input", "label", "qc")
)
samples <- samples %>%
unite(label, treat, !!sym(rep_col), remove = FALSE) %>%
mutate(
treat = factor(treat, levels = treat_levels),
"{rep_col}" := as.factor(!!sym(rep_col))
) %>%
dplyr::filter(treat %in% treat_levels)
stopifnot(nrow(samples) > 0)## Seqinfo
sq <- read_rds(file.path(annotation_path, "seqinfo.rds"))
## Blacklist
blacklist <- import.bed(here::here(config$external$blacklist), seqinfo = sq)
## Gene Annotations
gtf_gene <- read_rds(file.path(annotation_path, "gtf_gene.rds"))
gtf_transcript <- read_rds(file.path(annotation_path, "gtf_transcript.rds"))
external_features <- c()
has_features <- FALSE
if (!is.null(config$external$features)) {
external_features <- suppressWarnings(
import.gff(here::here(config$external$features), genome = sq)
)
keep_cols <- !vapply(
mcols(external_features), function(x) all(is.na(x)), logical(1)
)
mcols(external_features) <- mcols(external_features)[keep_cols]
has_features <- TRUE
}
gene_regions <- read_rds(file.path(annotation_path, "gene_regions.rds"))
regions <- vapply(gene_regions, function(x) unique(x$region), character(1))
rna_path <- here::here(config$external$rnaseq)
rnaseq <- tibble(gene_id = character())
if (length(rna_path) > 0) {
stopifnot(file.exists(rna_path))
if (str_detect(rna_path, "tsv$")) rnaseq <- read_tsv(rna_path)
if (str_detect(rna_path, "csv$")) rnaseq <- read_csv(rna_path)
if (!"gene_id" %in% colnames(rnaseq)) stop("Supplied RNA-Seq data must contain the column 'gene_id'")
gtf_gene <- subset(gtf_gene, gene_id %in% rnaseq$gene_id)
}
tx_col <- intersect(c("tx_id", "transcript_id"), colnames(rnaseq))
rna_gr_col <- ifelse(length(tx_col) > 0, "transcript_id", "gene_id")
rna_col <- c(tx_col, "gene_id")[[1]]
tss <- read_rds(file.path(annotation_path, "tss.rds"))
## bands_df
cb <- config$genome$build %>%
str_to_lower() %>%
paste0(".cytobands")
data(list = cb)
bands_df <- get(cb)fig_path <- here::here("docs", "assets", target)
if (!dir.exists(fig_path)) dir.create(fig_path, recursive = TRUE)
fig_dev <- knitr::opts_chunk$get("dev")
fig_type <- fig_dev[[1]]
if (is.null(fig_type)) stop("Couldn't detect figure type")
fig_fun <- match.fun(fig_type)
if (fig_type %in% c("bmp", "jpeg", "png", "tiff")) {
## These figure types require dpi & resetting to be inches
formals(fig_fun)$units <- "in"
formals(fig_fun)$res <- 300
}bfl <- bam_path %>%
file.path(glue("{samples$sample}.bam")) %>%
BamFileList() %>%
setNames(samples$sample)individual_peaks <- file.path(
macs2_path, samples$sample, glue("{samples$sample}_peaks.narrowPeak")
) %>%
importPeaks(seqinfo = sq, blacklist = blacklist) %>%
endoapply(names_to_column, var = "sample") %>%
endoapply(mutate, sample = str_remove(sample, "_peak_[0-9]+$")) %>%
endoapply(
mutate,
treat = left_join(tibble(sample = sample), samples, by = "sample")$treat
) %>%
setNames(samples$sample)macs2_logs <- macs2_path %>%
file.path(samples$sample, glue("{samples$sample}_callpeak.log") ) %>%
importNgsLogs() %>%
dplyr::select(
-contains("file"), -outputs, -n_reads, -alt_fragment_length
) %>%
left_join(samples, by = c("name" = "sample")) %>%
mutate(
total_peaks = map_int(
name, function(x) length(individual_peaks[[x]])
)
)
n_reps <- macs2_logs %>%
group_by(treat) %>%
summarise(n = sum(qc == "pass"))QC
This section provides a simple series of visualisations to enable detection of any problematic samples.
- Library Sizes: These are the total number of alignments contained in
each
bamfile, as passed tomacs2 callpeak(Zhang et al. 2008) - GC Content: Most variation in GC-content should have been identified
prior to performing alignments, using common tools such as FastQC, MultiQC (Ewels et al. 2016) or
ngsReports(Ward, To, and Pederson 2019). However, these plots may still be informative for detection of potential sequencing issues not previously addressed - Peaks Detected: The number of peaks detected within each individual
replicate are shown here, and provide clear guidance towards any samples
where the IP may have been less successful, or there may be possible
sample mis-labelling. Using the settings provided in
config.yml(i.e.peaks:qc:outlier_threshold), any replicates where the number of peaks falls outside the range defined by \(\pm\) 10-fold of the median peak number within each treatment group will be marked as failing QC. Whilst most cell-line generated data-sets are consistent, organoid or solid-tissue samples are far more prone to high variability in the success of the IP step. - Cross Correlations: Shows the cross-correlation coefficients between read positions across a series of intervals (Lun and Smyth 2014). Weak cross-correlations can also indicate low-quality samples. These values are also used to estimate fragment length within each sample, as the peak value of the cross-correlations
- Fraction Of Reads In Peaks (FRIP): This plot shows the proportion of
the alignments which fall within peaks identified by
macs2 callpeak, with the remainder of alignments being assumed to be background (Landt et al. 2012). This can provide guidance as to the success of the IP protocol, and the common-use threshold of 1% is indicated as a horizontal line. This value is not enforced as a hard QC criteria, but may be used to manually exclude samples from the filesamples.tsvof deemed to be appropriate.
emphasize.italics.rows(NULL)
any_fail <- any(macs2_logs$qc == "fail")
if (any_fail) emphasize.italics.rows(which(macs2_logs$qc == "fail"))
macs2_logs %>%
dplyr::select(
sample = name, label,
total_peaks,
reads = n_tags_treatment, read_length = tag_length,
fragment_length
) %>%
rename_all(str_sep_to_title )%>%
pander(
justify = "llrrrr",
caption = glue(
"*Summary of results for `macs2 callpeak` on individual {target} samples.",
"Total peaks indicates the number retained after applying the FDR ",
"threshold of {percent(config$peaks$macs2$fdr)} during the peak calling ",
"process.",
ifelse(
any_fail,
glue(
"Samples shown in italics were marked for exclusion from ",
"downstream analysis as they identified a number of peaks beyond ",
"+/- {config$peaks$qc$outlier_threshold}-fold the median number of ",
"peaks amongst all samples within the relevant treatment group.",
),
glue(
"No samples were identified as failing QC based on the number of ",
"peaks identified relative to the highest quality sample."
)
),
"Any peaks passing the FDR cutoff, but which overlapped any black-listed",
"regions were additionally excluded. The fragment length as estimated by",
"`macs2 predictd` is given in the final column.",
case_when(
all(macs2_logs$paired_end) ~
"All input files contained paired-end reads.*",
all(!macs2_logs$paired_end) ~
"All input files contained single-end reads.*",
TRUE ~
"Input files were a mixture of paired and single-end reads*"
),
.sep = " "
)
)| Sample | Label | Total Peaks | Reads | Read Length | Fragment Length |
|---|---|---|---|---|---|
| SRR8315180 | E2_1 | 5,663 | 16,373,508 | 68 | 155 |
| SRR8315181 | E2_2 | 7,722 | 17,581,871 | 65 | 175 |
| SRR8315182 | E2_3 | 12,084 | 17,641,037 | 73 | 176 |
| SRR8315183 | E2DHT_1 | 6,290 | 16,208,256 | 66 | 167 |
| SRR8315184 | E2DHT_2 | 10,989 | 18,270,619 | 73 | 178 |
| SRR8315185 | E2DHT_3 | 7,371 | 18,061,306 | 68 | 179 |
Library Sizes
macs2_logs %>%
ggplot(
aes(label, n_tags_treatment, fill = qc)
) +
geom_col(position = "dodge") +
geom_hline(
aes(yintercept = mn),
data = . %>%
group_by(treat) %>%
summarise(mn = mean(n_tags_treatment)),
linetype = 2,
col = "grey"
) +
facet_grid(~treat, scales = "free_x", space = "free_x") +
scale_y_continuous(expand = expansion(c(0, 0.05)), labels = comma) +
scale_fill_manual(values = colours$qc) +
labs(
x = "Sample",
y = "Library Size",
fill = "QC"
) +
ggtitle(
glue("{target}: Library Sizes")
)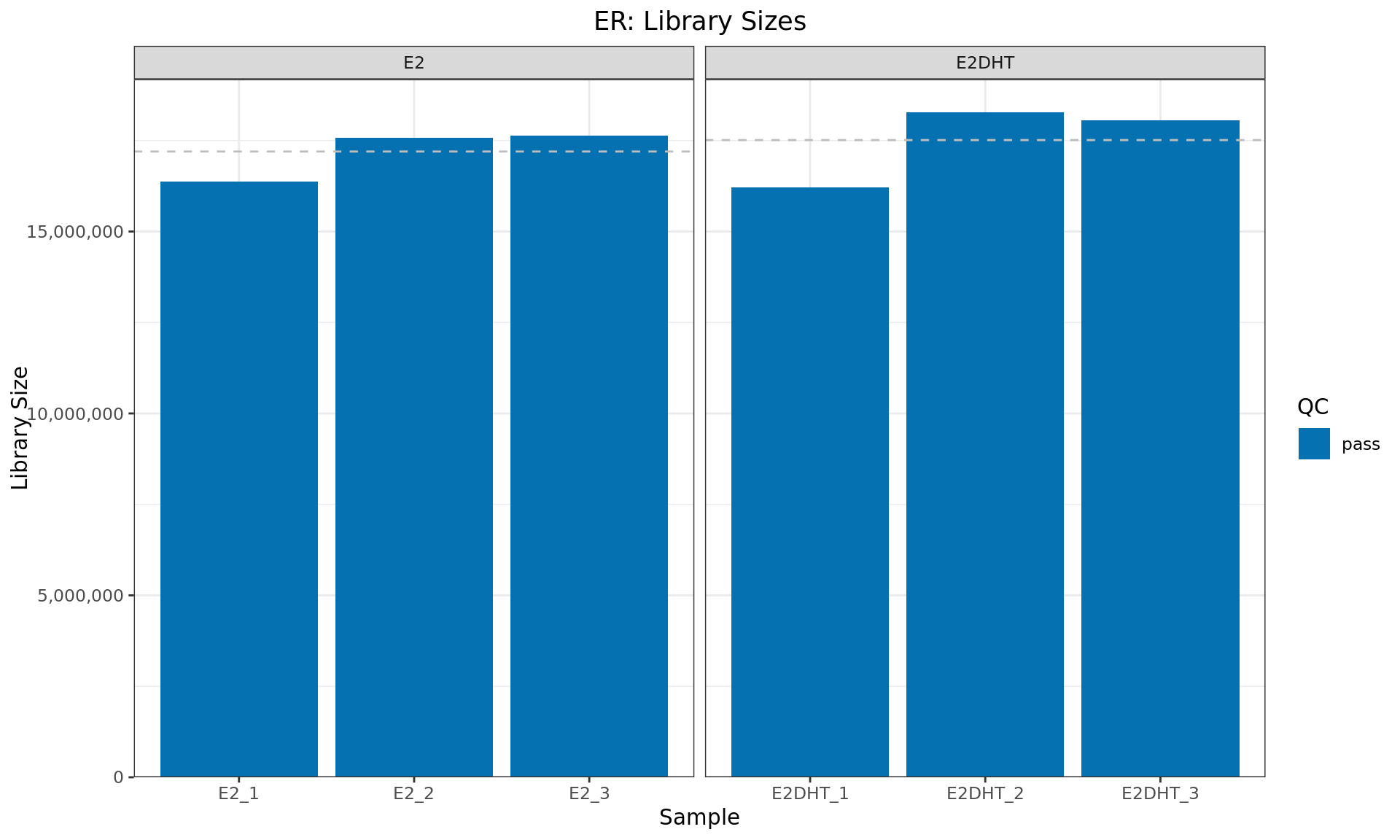
Library sizes for each ER sample. The horizontal line indicates the mean library size for each treatment group. Any samples marked for exclusion as described above will be indicated with an (F)
Peaks Detected
suppressWarnings(
macs2_logs %>%
ggplot(aes(label, total_peaks, fill = qc)) +
geom_col() +
geom_label(
aes(x = label, y = total_peaks, label = lab, colour = qc),
data = . %>%
dplyr::filter(total_peaks > 0) %>%
mutate(
lab = comma(total_peaks, accuracy = 1), total = total_peaks
),
nudge_y = 0.03 * max(macs2_logs$total_peaks),
inherit.aes = FALSE, show.legend = FALSE
) +
facet_grid(~treat, scales = "free_x", space = "free_x") +
scale_y_continuous(expand = expansion(c(0, 0.05)), labels = comma) +
scale_fill_manual(values = colours$qc) +
scale_colour_manual(values = colours$qc) +
labs(
x = "Sample", y = "Total Peaks", fill = "QC"
) +
ggtitle(glue("{target}: Number of Peaks"))
)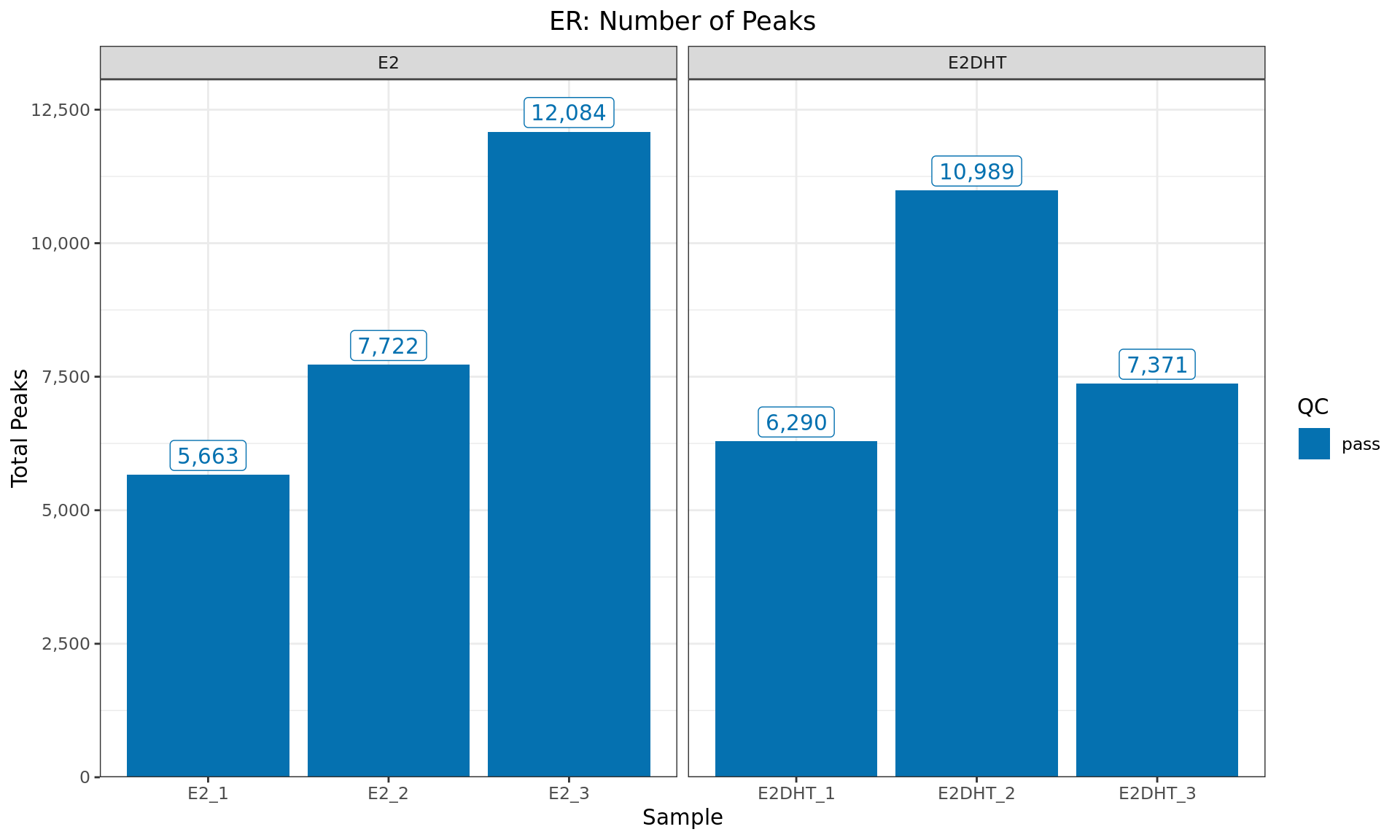
Peaks identified for each ER sample. The number of peaks passing the
inclusion criteria for macs2 callpeak (FDR < 0.05) are
provided. Any samples marked for exclusion are coloured as indicated in
the figure legend.
FRIP
samples$sample %>%
bplapply(
function(x) {
gr <- individual_peaks[[x]]
rip <- 0
if (length(gr) > 0) {
sbp <- ScanBamParam(which = gr)
rip <- sum(countBam(bfl[[x]], param = sbp)$records)
}
tibble(name = x, reads_in_peaks = rip)
}
) %>%
bind_rows() %>%
left_join(macs2_logs, by = "name") %>%
mutate(frip = reads_in_peaks / n_tags_treatment) %>%
ggplot(aes(label, frip, fill = qc)) +
geom_col() +
geom_hline(yintercept = 0.01, colour = "grey", linetype = 2) +
facet_grid(~treat, scales = "free_x", space = "free") +
scale_y_continuous(labels = percent, expand = expansion(c(0, 0.05))) +
scale_fill_manual(values = colours$qc) +
labs(
x = "Sample", y = "Fraction of Reads In Peaks", fill = "QC"
) +
ggtitle(glue("{target}: Fraction Of Reads In Peaks"))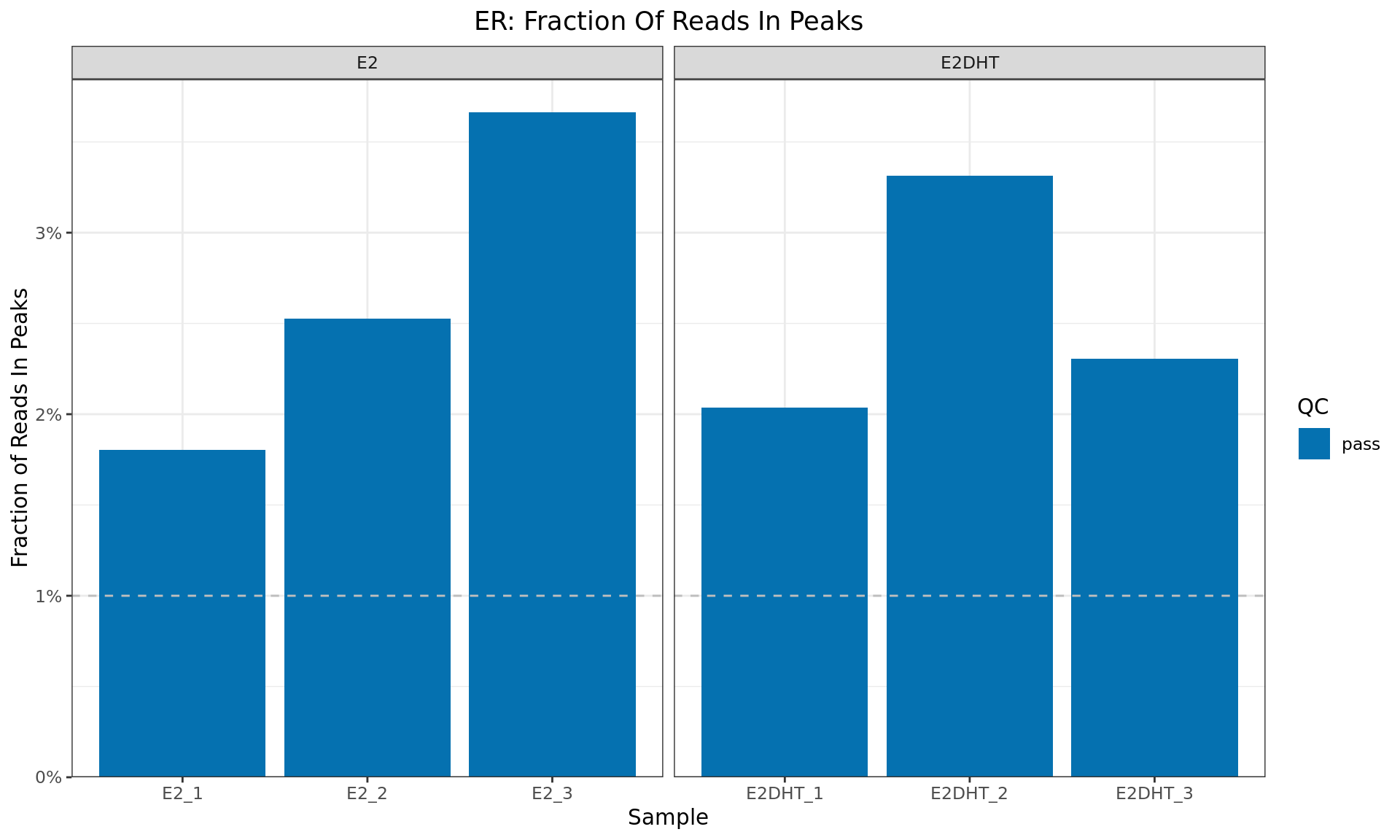
Fraction of Reads In Peaks for each sample. Higher values indicate more reads specifically associated with the ChIP target (ER). The common-use minimum value for an acceptable sample (1%) is shown as a dashed horizontal line
Cross Correlations
macs2_path %>%
file.path(target, glue("{target}_cross_correlations.tsv")) %>%
read_tsv() %>%
left_join(samples, by = "sample") %>%
ggplot(aes(fl, correlation, colour = treat)) +
geom_point(alpha = 0.1) +
geom_smooth(se = FALSE, method = 'gam', formula = y ~ s(x, bs = "cs")) +
geom_vline(
aes(xintercept = fragment_length),
data = macs2_logs,
colour = "grey40", linetype = 2
) +
geom_label(
aes(label = fl),
data = . %>%
mutate(correlation = correlation + 0.03 * max(correlation)) %>%
dplyr::filter(correlation == max(correlation), .by = sample),
show.legend = FALSE, alpha = 0.7
) +
facet_grid(as.formula(paste("treat ~", rep_col))) +
scale_colour_manual(values = colours$treat[treat_levels]) +
scale_x_continuous(
breaks = seq(0, 5*max(macs2_logs$fragment_length), by = 200)
) +
labs(
x = "Distance (bp)", y = "Cross Correlation", colour = "Treat"
) +
ggtitle(glue("{target}: Cross Correlations"))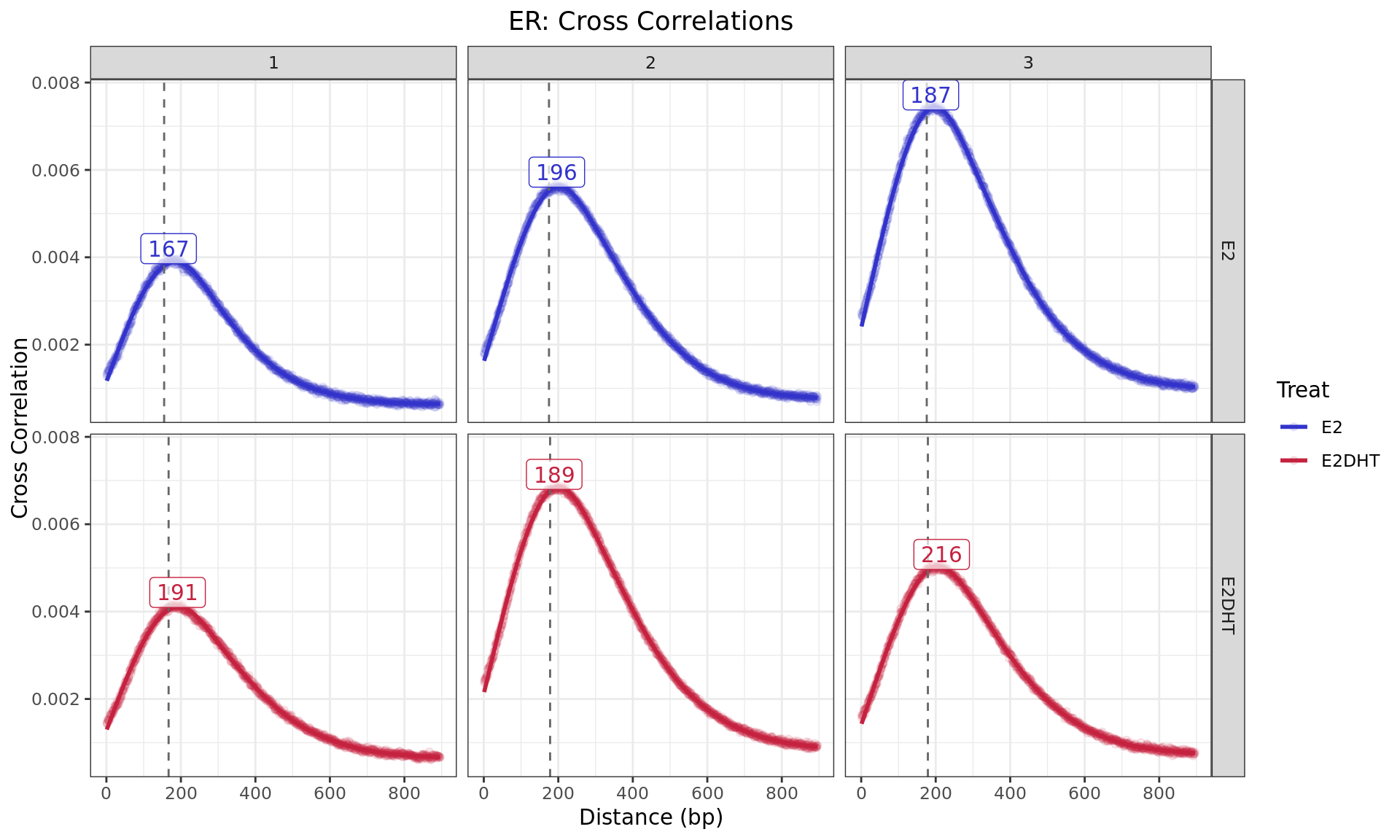
Cross Correlaton between alignments up to 1kb apart. The dashed,
grey, vertical line is the fragment length estimated by
macs2 callpeak for each sample, with labels indicating the
approximate point of the highest correlation, as representative of the
average fragment length. For speed, only the first 5 chromosomes were
used for sample-specific estimates.
GC Content
ys <- 5e5
yieldSize(bfl) <- ysbfl %>%
bplapply(
function(x){
seq <- scanBam(x, param = ScanBamParam(what = "seq"))[[1]]$seq
freq <- letterFrequency(seq, letters = "GC") / width(seq)
list(freq[,1])
}
) %>%
as_tibble() %>%
pivot_longer(
cols = everything(),
names_to = "name",
values_to = "freq"
) %>%
left_join(macs2_logs, by = "name") %>%
unnest(freq) %>%
ggplot(aes(label, freq, fill = qc)) +
geom_boxplot(alpha = 0.8) +
geom_hline(
aes(yintercept = med),
data = . %>%
group_by(treat) %>%
summarise(med = median(freq)),
linetype = 2,
colour = rgb(0.2, 0.2, 0.8)
) +
facet_grid(~treat, scales = "free_x", space = "free_x") +
scale_y_continuous(labels = percent) +
scale_fill_manual(values = colours$qc) +
labs(
x = "Sample",
y = "GC content",
fill = "QC"
) +
ggtitle(
glue("{target}: GC Content")
)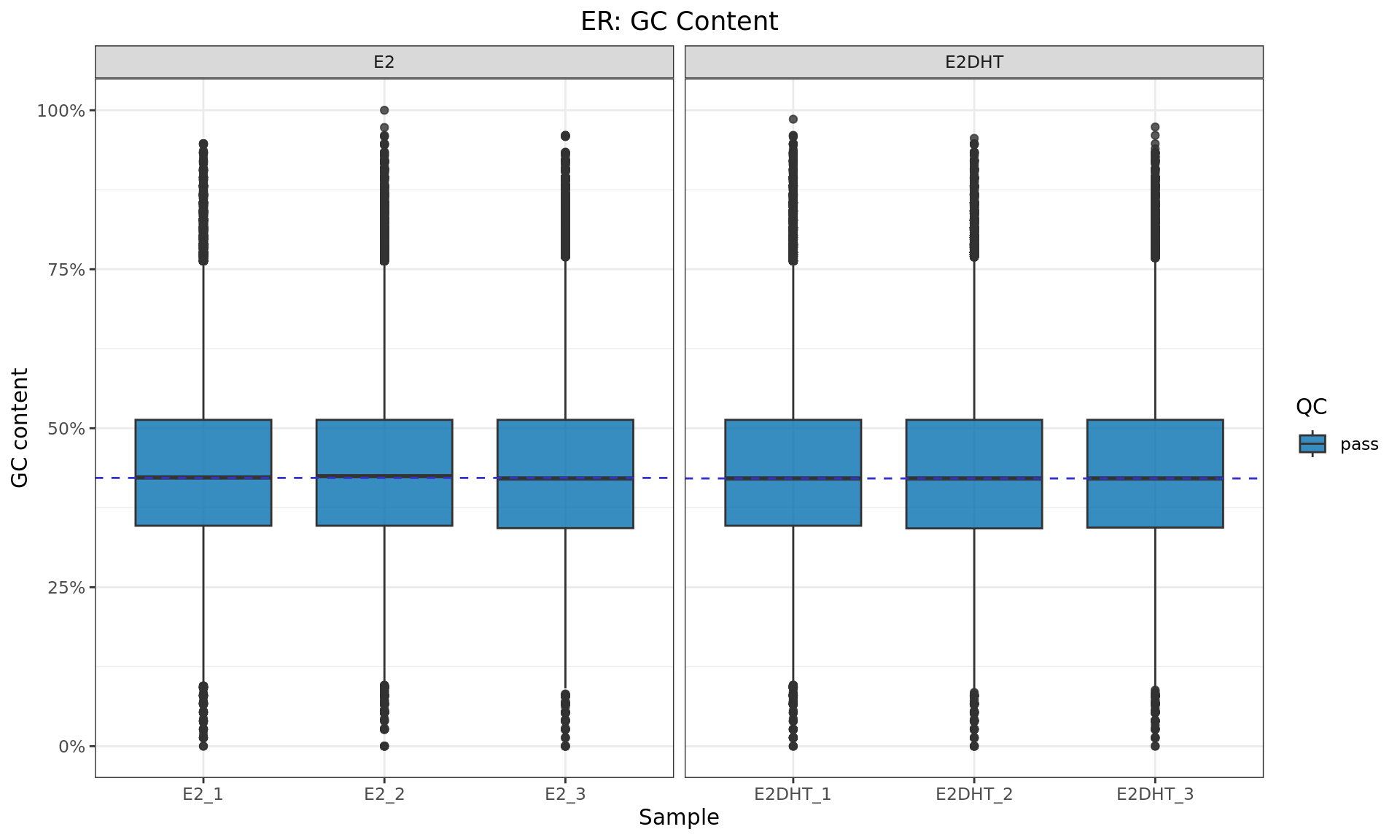
GC content for each bam file, taking the first 500,000 alignments from each sample. QC status is based on the number of peaks identified (see table above)
Results
Individual Replicates
Replicates within treatment groups are only shown where at least one
peak was detected. For two or three replicates, Euler (Venn) Diagrams
were generated using the python package
matplotlib-venn. Peak numbers may differ from above as
those shown below are overlapping peaks which have been merged across
replicates
In the case of four or more replicates, the package
ComplexUpset was used to create UpSet plots (Lex et
al. 2014), with colours indicating QC status.
All Samples
sample2label <- setNames(samples$label, samples$sample)
sets <- list(all = paste(samples$label, collapse = "; ")) %>%
c(
samples %>%
split(.$treat) %>%
lapply(function(x) paste(x$label, collapse = "; "))
) %>%
unlist()
valid_sets <- makeConsensus(individual_peaks) %>%
as_tibble() %>%
pivot_longer(
cols = all_of(samples$sample), names_to = "sample", values_to = "has_peak"
) %>%
dplyr::filter(has_peak) %>%
mutate(sample = sample2label[sample]) %>%
summarise(
samples = paste(sample, collapse = "; "), .by = range
) %>%
distinct(samples) %>%
dplyr::filter(samples %in% sets) %>%
pull(samples) %>%
setNames(vapply(., function(x){names(which(sets == x))}, character(1))) %>%
lapply(function(x) str_split(x, "; ")[[1]])
## Use upset_query objects to fill key intersections and sets
ql <- samples$label %>%
lapply(
function(x) upset_query(
set = x,
fill = colours$treat[as.character(dplyr::filter(samples, label == x)$treat)]
)
)
if (length(valid_sets) > 0) {
ql <- c(
ql,
names(valid_sets) %>%
lapply(
function(x) {
col <- ifelse(x == "all", "darkorange", colours$treat[[x]])
upset_query(
intersect = valid_sets[[x]],
only_components = "intersections_matrix",
color = col, fill = col
)
}
)
)
}
lb <- rev(arrange(samples, treat)$label)
size <- get_size_mode('exclusive_intersection')
individual_peaks %>%
setNames(sample2label[names(.)]) %>%
.[lb] %>%
plotOverlaps(
var = "qValue", f = "median",
.sort_sets = FALSE, queries = ql,
base_annotations = list(
`Peaks in Intersection` = intersection_size(
text_mapping = aes(label = comma(!!size)),
bar_number_threshold = 1, text_colors = "black",
text = list(size = 3.5, angle = 90, vjust = 0.5, hjust = -0.1)
) +
scale_y_continuous(expand = expansion(c(0, 0.25)), label = comma) +
theme(
panel.grid = element_blank(),
axis.line = element_line(colour = "grey20"),
panel.border = element_rect(colour = "grey20", fill = NA)
)
),
annotations = list(
qValue = ggplot(mapping = aes(y = qValue)) +
geom_boxplot(na.rm = TRUE, outlier.colour = rgb(0, 0, 0, 0.2)) +
coord_cartesian(
ylim = c(0, quantile(unlist(individual_peaks)$qValue, 0.99))
) +
scale_y_continuous(expand = expansion(c(0, 0.05))) +
ylab(expr(paste("Macs2 ", q[med]))) +
theme(
panel.grid = element_blank(),
axis.line = element_line(colour = "grey20"),
panel.border = element_rect(colour = "grey20", fill = NA)
)
),
set_sizes = (
upset_set_size() +
geom_text(
aes(label = comma(after_stat(count))),
hjust = 1.1, stat = 'count', size = 3.5
) +
scale_y_reverse(expand = expansion(c(0.3, 0)), label = comma) +
ylab(glue("Macs2 Peaks (FDR < {config$peaks$macs2$fdr})")) +
theme(
panel.grid = element_blank(),
axis.line = element_line(colour = "grey20"),
panel.border = element_rect(colour = "grey20", fill = NA)
)
),
min_size = 10, n_intersections = 40
) +
theme(
panel.grid = element_blank(),
panel.border = element_rect(colour = "grey20", fill = NA),
axis.line = element_line(colour = "grey20")
) +
labs(x = "Intersection") +
patchwork::plot_layout(heights = c(2, 3, 2))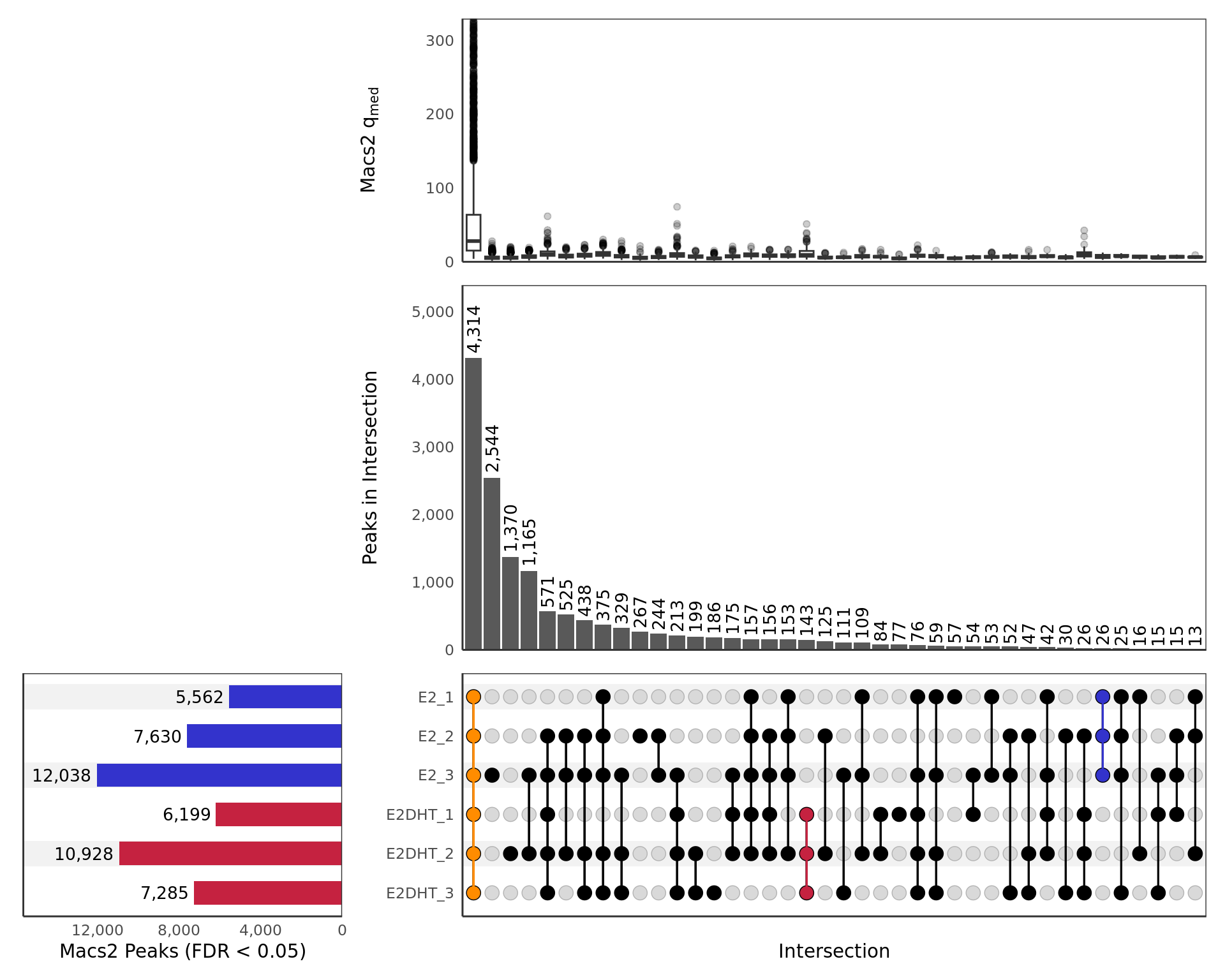
UpSet plot showing all samples including those which failed prior QC
steps. Any potential sample mislabelling will show up clearly here as
samples from each group should show a preference to overlap other
samples within the same treatment group. Intersections are only included
if 10 or more sites are present. The top panel shows a boxplot of the
median \(q\)-values produced by
macs2 callpeak for each peak in the intersection. The
y-axis for this panel is truncated at the 99th percentile of
values. Only intersections with 10 or more peaks are shown.
ranges_by_rep_treat <- individual_peaks %>%
unlist() %>%
splitAsList(.$treat) %>%
.[vapply(., length, integer(1)) > 1] %>%
lapply(select, sample) %>%
lapply(reduceMC) %>%
lapply(as_tibble) %>%
lapply(unnest, sample) %>%
lapply(mutate, detected = 1) %>%
lapply(left_join, dplyr::select(samples, sample, label), by = "sample") %>%
lapply(dplyr::select, -sample) %>%
lapply(function(x) {
split(x, x$label) %>%
lapply(pull, "range")
}
)
## First find the python executable
which_py <- c(
file.path(Sys.getenv("CONDA_PREFIX"), "bin", "python3"),
Sys.which("python3")
) %>%
.[file.exists(.)] %>%
.[1]
stopifnot(length(which_py) == 1)
rep_col <- hcl.colors(3, palette = "Zissou", rev = TRUE)
htmltools::tagList(
names(ranges_by_rep_treat) %>%
lapply(
function(i) {
x <- ranges_by_rep_treat[[i]]
fig_out <- file.path(fig_path, glue("{i}_replicates.{fig_type}"))
if (length(x) == 1) {
fig_fun(
fig_out,
width = knitr::opts_chunk$get("fig.width"),
height = ifelse(
length(x) < 3,
knitr::opts_chunk$get("fig.height") * 0.8,
knitr::opts_chunk$get("fig.height") * 0.8
)
)
grid.newpage()
draw.single.venn(length(x[[1]]), category = names(x))
dev.off()
}
## Euler diagrams for 2-3 replicates.
if (length(x) == 2) {
a12 <- sum(duplicated(unlist(x)))
a1 <- length(x[[1]]) - a12
a2 <- length(x[[2]]) - a12
args <- glue(
here::here("workflow", "scripts", "plot_venn.py "),
"-a1 {a1} -a2 {a2} -a12 {a12} ",
"-s1 '{names(x)[[1]]}' -s2 '{names(x)[[2]]}' ",
"-c1 '{rep_col[[1]]}' -c2 '{rep_col[[3]]}' ",
"-ht {knitr::opts_chunk$get('fig.height')} ",
"-w {knitr::opts_chunk$get('fig.width')} ",
"-o '{fig_out}'"
)
system2(which_py, args)
}
if (length(x) == 3) {
a123 <- sum(table(unlist(x)) == 3)
a12 <- sum(table(unlist(x[1:2])) == 2) - a123
a13 <- sum(table(unlist(x[c(1, 3)])) == 2) - a123
a23 <- sum(table(unlist(x[c(2, 3)])) == 2) - a123
a1 <- length(x[[1]]) - (a12 + a13 + a123)
a2 <- length(x[[2]]) - (a12 + a23 + a123)
a3 <- length(x[[3]]) - (a13 + a23 + a123)
args <- glue(
here::here("workflow", "scripts", "plot_venn.py "),
"-a1 {a1} -a2 {a2} -a12 {a12} -a3 {a3} ",
"-a13 {a13} -a23 {a23} -a123 {a123} ",
"-s1 '{names(x)[[1]]}' -s2 '{names(x)[[2]]}' -s3 '{names(x)[[3]]}' ",
"-c1 '{rep_col[[1]]}' -c2 '{rep_col[[2]]}' -c3 '{rep_col[[3]]}' ",
"-ht {knitr::opts_chunk$get('fig.height')} ",
"-w {knitr::opts_chunk$get('fig.width')} ",
"-o '{fig_out}'"
)
system2(which_py, args)
}
## An UpSet plot for > 3 replicates
if (length(x) > 3) {
fig_fun(
fig_out,
width = knitr::opts_chunk$get("fig.width"),
height = knitr::opts_chunk$get("fig.height")
)
grid.newpage()
## Prep the data for ComplexUpset
df <- individual_peaks[dplyr::filter(samples, treat == i)$sample] %>%
makeConsensus(var = "qValue") %>%
mutate(qValue = vapply(qValue, median, numeric(1))) %>%
as_tibble() %>%
dplyr::rename_with(
function(x) setNames(samples$label, samples$sample)[x],
any_of(samples$sample)
)
## Highlight samples by QC
ql <- samples %>%
dplyr::filter(treat == i) %>%
pull('label') %>%
lapply(
function(x) {
i <- as.character(
dplyr::filter(samples, treat == i, label == x)$qc
)
upset_query(set = x, fill = colours$qc[i])
}
)
## And plot
p <- df %>%
upset(
intersect = rev(dplyr::filter(samples, treat == i)$label),
base_annotations = list(
`Peaks in Intersection` = intersection_size(
text_mapping = aes(label = comma(!!size)),
bar_number_threshold = 1, text_colors = "black",
text = list(size = 4, hjust = 0.5)
) +
scale_y_continuous(expand = expansion(c(0, 0.2)), label = comma) +
theme(
text = element_text(size = 14),
panel.grid = element_blank(),
axis.line = element_line(colour = "grey20"),
panel.border = element_rect(colour = "grey20", fill = NA)
)
),
annotations = list(
qValue = ggplot(mapping = aes(y = qValue)) +
geom_boxplot(na.rm = TRUE, outlier.colour = rgb(0, 0, 0, 0.2)) +
coord_cartesian(ylim = c(0, quantile(df$qValue, 0.99))) +
scale_y_continuous(expand = expansion(c(0, 0.05))) +
ylab(expr(paste("Macs2 ", q[med]))) +
theme(
text = element_text(size = 14),
panel.grid = element_blank(),
axis.line = element_line(colour = "grey20"),
panel.border = element_rect(colour = "grey20", fill = NA)
)
),
set_sizes = (
upset_set_size() +
geom_text(
aes(label = comma(after_stat(count))),
hjust = 1.1, stat = 'count', size = 4
) +
scale_y_reverse(expand = expansion(c(0.2, 0)), label = comma) +
ylab(glue("Macs2 Peaks (FDR < {config$peaks$macs2$fdr})")) +
theme(
text = element_text(size = 14),
panel.grid = element_blank(),
axis.line = element_line(colour = "grey20"),
panel.border = element_rect(colour = "grey20", fill = NA)
)
),
queries = ql,
min_size = 10,
sort_sets = FALSE
) +
theme(text = element_text(size = 14)) +
patchwork::plot_layout(heights = c(2, 3, 2))
print(p)
dev.off()
}
## Define the caption
cp <- htmltools::tags$em(
glue(
"
Number of {target} peaks detected by macs2 callpeak in each {i}
replicate and the number of peaks shared between replicates.
Replicates which passed/failed QC are show in the specified colours.
"
)
)
## Create html tags
fig_link <- str_extract(fig_out, "assets.+")
htmltools::div(
htmltools::div(
id = glue("{i}-replicates"),
class = "section level4",
htmltools::h4(i),
htmltools::div(
class = "figure", style = "text-align: center",
htmltools::img(src = fig_link, width = 960),
htmltools::p(
class = "caption", htmltools::tags$em(cp)
)
)
)
)
}
)
)E2
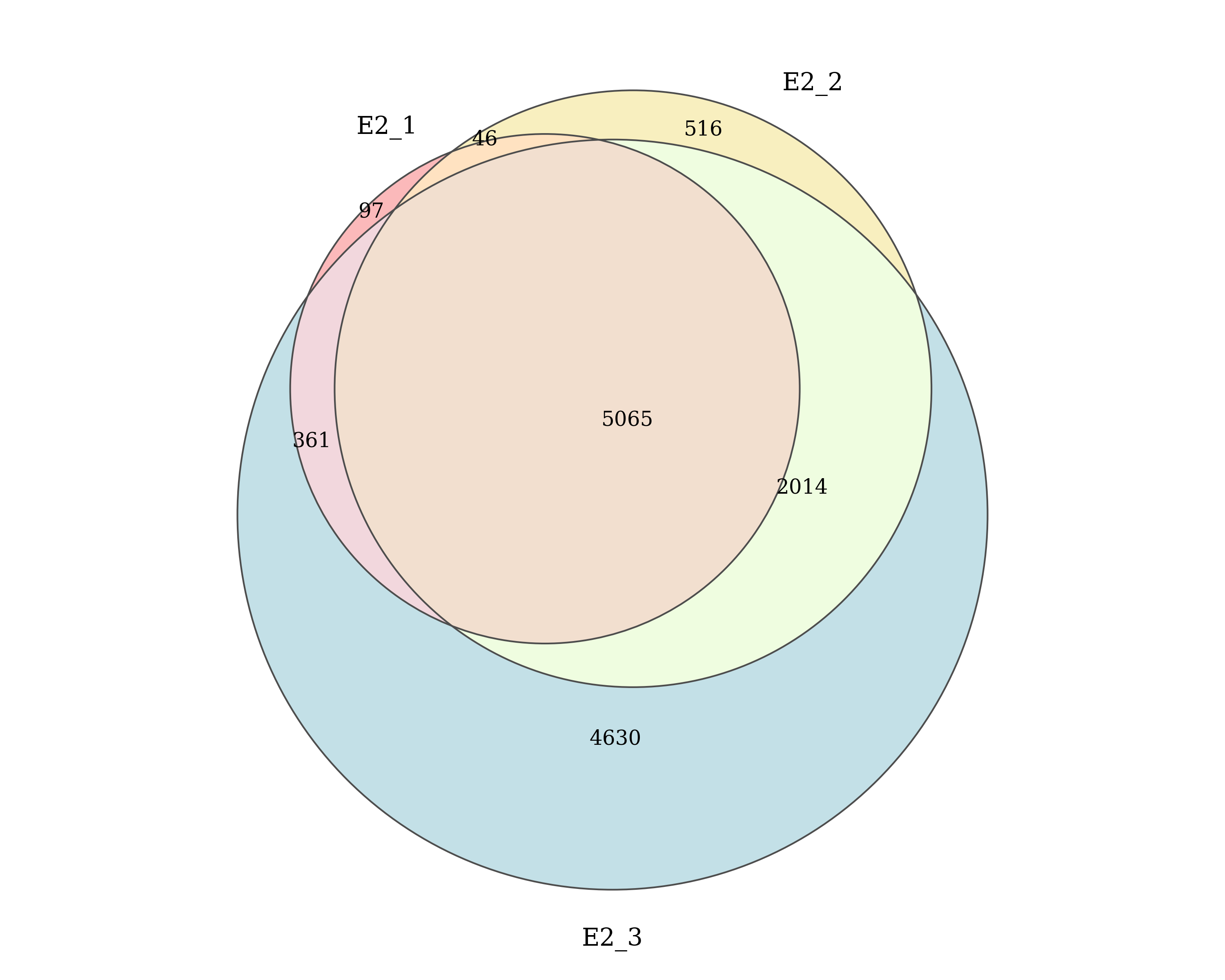
Number of ER peaks detected by macs2 callpeak in each E2 replicate and the number of peaks shared between replicates. Replicates which passed/failed QC are show in the specified colours.
E2DHT
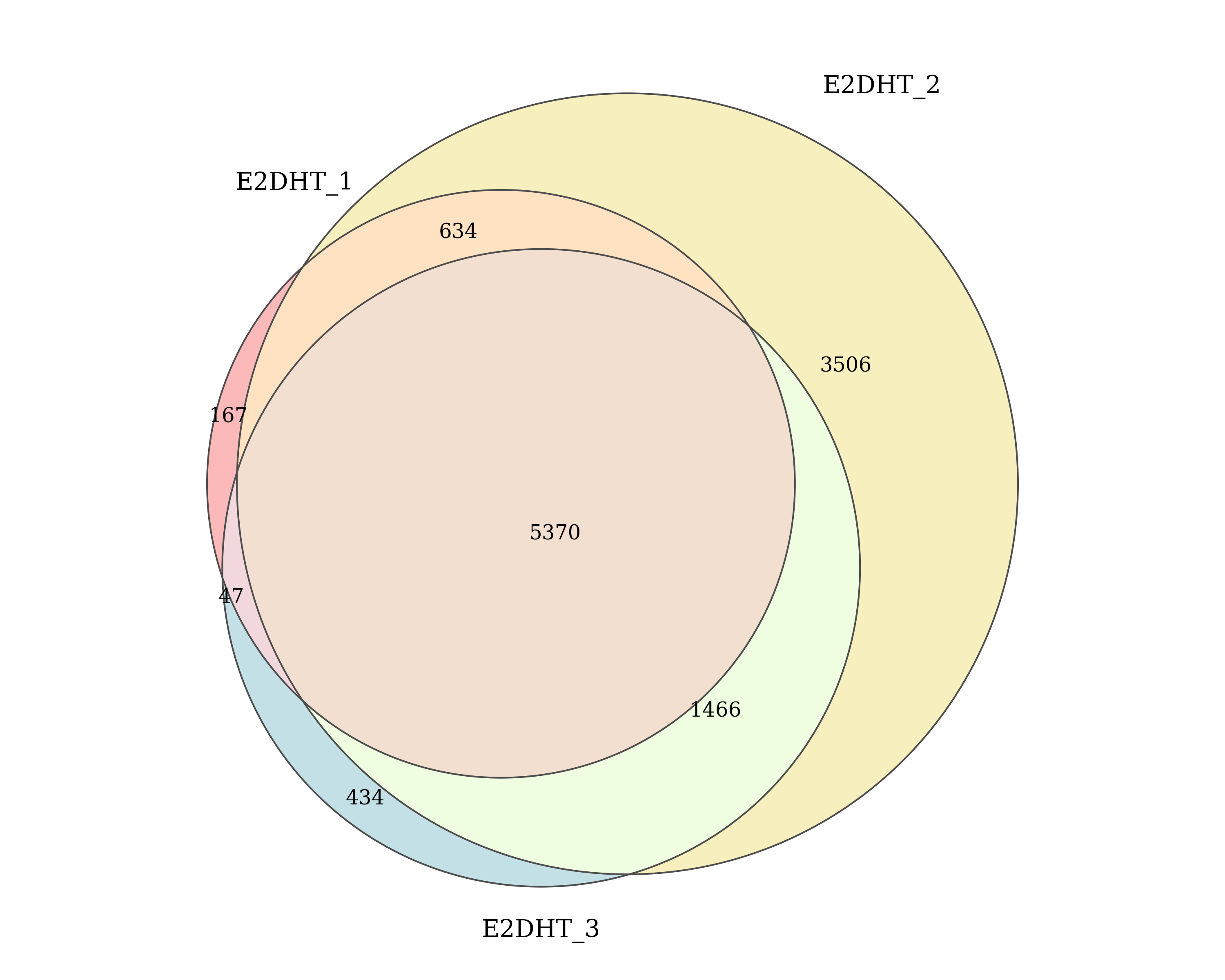
Number of ER peaks detected by macs2 callpeak in each E2DHT replicate and the number of peaks shared between replicates. Replicates which passed/failed QC are show in the specified colours.
Individual Peak Widths
individual_peaks %>%
width() %>%
as.list() %>%
lapply(list) %>%
as_tibble() %>%
pivot_longer(cols = everything(), names_to = "sample", values_to = "width") %>%
unnest(width) %>%
left_join(samples, by = "sample") %>%
ggplot(aes(label, width, fill = treat)) +
geom_boxplot(alpha = 0.9) +
geom_hline(
yintercept = median(width(unlist(individual_peaks))),
linetype = 2
) +
facet_wrap(~treat, nrow = 1, scales = "free_x") +
scale_y_log10() +
scale_fill_manual(values = colours$treat) +
labs(x = "Sample", y = "Peak Width (bp)", fill = "Treatment")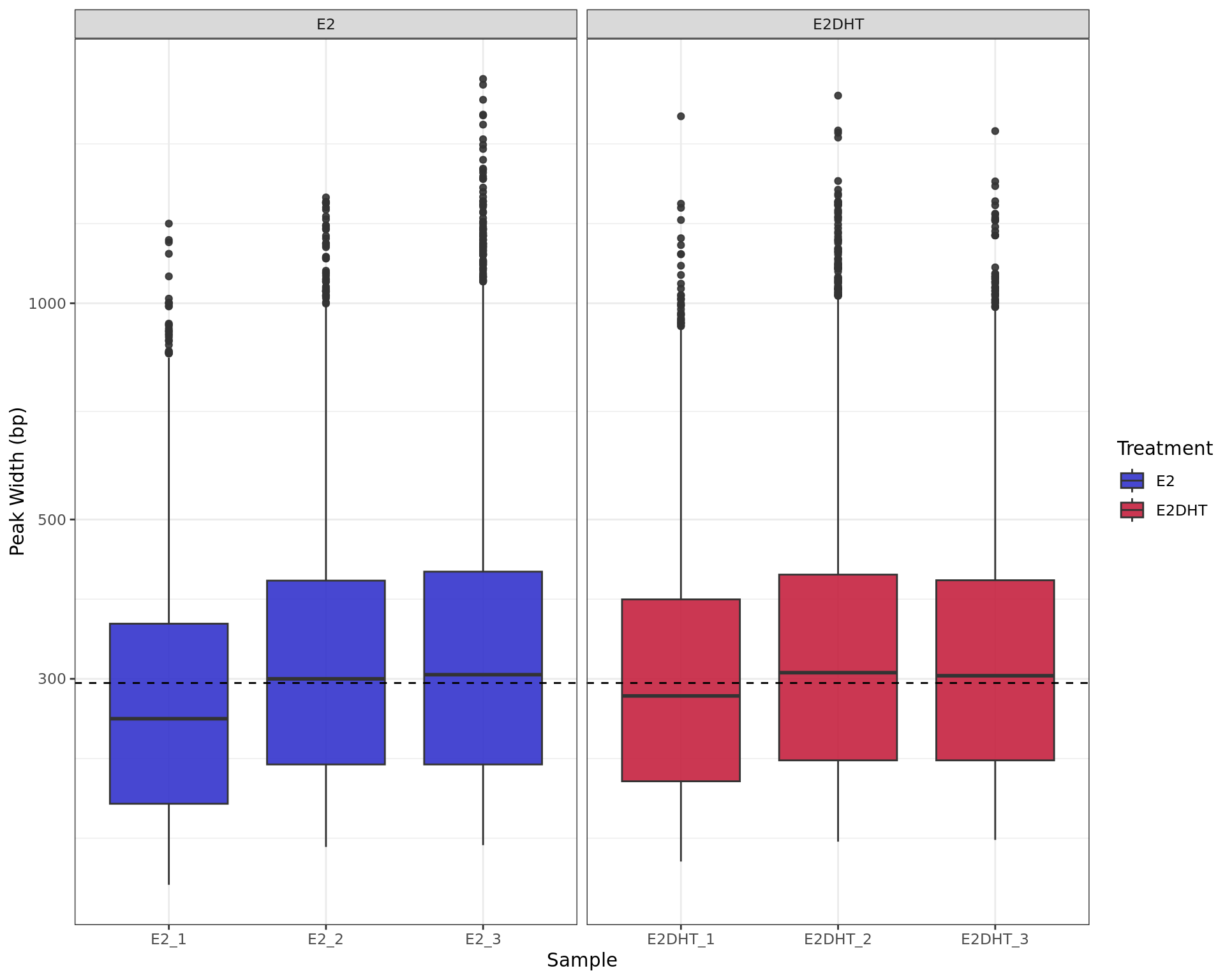
Range of peak widths across all samples. The median value for all peaks (296bp) is shown as the dashed horizontal line
Treatment Peaks
treatment_peaks <- treat_levels %>%
sapply(
function(x) {
gr <- macs2_path %>%
file.path(target, glue("{target}_{x}_merged_peaks.narrowPeak")) %>%
importPeaks(seqinfo = sq, blacklist = blacklist, centre = TRUE) %>%
unlist()
k <- dplyr::filter(n_reps, treat == x)$n * config$peaks$qc$min_prop_reps
if (k > 0) {
samp <- dplyr::filter(macs2_logs, treat == x, qc != "fail")$name
gr$n_reps <- countOverlaps(gr, individual_peaks[samp])
gr$keep <- gr$n_reps >= k
} else {
gr <- GRanges(seqinfo = sq)
}
gr
},
simplify = FALSE
) %>%
GRangesList()
union_peaks <- treatment_peaks %>%
endoapply(subset, keep) %>%
makeConsensus(
var = c("score", "signalValue", "pValue", "qValue", "centre")
) %>%
mutate(
score = vapply(score, median, numeric(1)),
signalValue = vapply(signalValue, median, numeric(1)),
pValue = vapply(pValue, median, numeric(1)),
qValue = vapply(qValue, median, numeric(1)),
centre = vapply(centre, median, numeric(1)),
region = bestOverlap(
.,
lapply(gene_regions, select, region) %>%
GRangesList() %>%
unlist(),
var = "region"
) %>%
factor(levels = regions)
)
if (has_features) {
union_peaks$Feature <- bestOverlap(
union_peaks, external_features,
var = "feature", missing = "no_feature"
) %>%
factor(levels = names(colours$features)) %>%
fct_relabel(str_sep_to_title)
}A set of treatment-specific ER peaks (i.e. treatment peaks) was defined for each condition by comparing the peaks detected when merging all treatment-specific samples, against those detected within each replicate. Replicates which failed the previous QC steps are omitted from this step. Treatment peaks which overlapped a peak in more than 50% of the individual replicates passing QC in each treatment group, were retained.
treatment_peaks %>%
lapply(
function(x) {
tibble(
detected_peaks = length(x),
retained = sum(x$keep),
`% retained` = percent(retained / detected_peaks, 0.1),
median_width = median(width(x))
)
}
) %>%
bind_rows(.id = "treat") %>%
mutate(treat = factor(treat, levels = treat_levels)) %>%
group_by(treat) %>%
rename_all(str_sep_to_title) %>%
dplyr::rename(`% Retained` = ` Retained`) %>%
pander(
justify = "lrrrr",
caption = glue(
"Treatment peaks detected by merging samples within each treatment group.",
"Peaks were only retained if detected in at least",
"{percent(config$peaks$qc$min_prop_reps)} of the retained samples for each", "treatment group, as described above.",
.sep = " "
)
)| Treat | Detected Peaks | Retained | % Retained | Median Width |
|---|---|---|---|---|
| E2 | 7,335 | 6,673 | 91.0% | 296 |
| E2DHT | 7,129 | 6,835 | 95.9% | 298 |
Union Peaks
vd <- treatment_peaks %>%
lapply(
function(x) {
as.character(
subsetByOverlaps(
union_peaks,
subset(x, keep)
)
)
}
) %>%
setNames(treat_levels) %>%
.[vapply(., length, integer(1)) > 0] In addition to the treatment peaks, a set of 7,532 treatment-agnostic
ER union peaks were defined. Union ranges were the
union of all overlapping ranges defined and retained in one or
more sets of treatment peaks. Resulting values for the
score, signalValue, pValue and
qValue were calculated as the median across all
treatments. Peak summits were taken as the median position of
all summits which comprise the union peak.
Venn Diagram
fig_name <- glue("{target}_common_peaks.{fig_type}")
fig_out <- file.path(fig_path, fig_name)
## An empty file to keep snakemake happy if > 3 treatments
file.create(fig_out)
if (length(vd) == 1) {
fig_fun(filename = fig_out, width = 8, height = 3)
grid.newpage()
draw.single.venn(
area = length(vd[[1]]), category = names(vd)[[1]],
fill = colours$treat[names(vd)], alpha = 0.3
)
dev.off()
}
if (length(vd) == 2) {
a1 <- length(setdiff(vd[[1]], vd[[2]]))
a2 <- length(setdiff(vd[[2]], vd[[1]]))
a12 <- sum(duplicated(unlist(vd)))
args <- glue(
here::here("workflow", "scripts", "plot_venn.py "),
"-a1 {a1} -a2 {a2} -a12 {a12} ",
"-s1 '{names(vd)[[1]]}' -s2 '{names(vd)[[2]]}' ",
"-c1 '{colours$treat[[names(vd)[[1]]]]}' ",
"-c2 '{colours$treat[[names(vd)[[2]]]]}' ",
"-ht {knitr::opts_chunk$get('fig.height')} ",
"-w {knitr::opts_chunk$get('fig.width')} ",
"-o '{fig_out}'"
)
system2(which_py, args)
}
if (length(vd) == 3) {
a123 <- sum(table(unlist(vd)) == 3)
a12 <- sum(table(unlist(vd[1:2])) == 2) - a123
a13 <- sum(table(unlist(vd[c(1, 3)])) == 2) - a123
a23 <- sum(table(unlist(vd[c(2, 3)])) == 2) - a123
a1 <- length(vd[[1]]) - (a12 + a13 + a123)
a2 <- length(vd[[2]]) - (a12 + a23 + a123)
a3 <- length(vd[[3]]) - (a13 + a23 + a123)
args <- glue(
here::here("workflow", "scripts", "plot_venn.py "),
"-a1 {a1} -a2 {a2} -a12 {a12} -a3 {a3} ",
"-a13 {a13} -a23 {a23} -a123 {a123} ",
"-s1 '{names(vd)[[1]]}' -s2 '{names(vd)[[2]]}' -s3 '{names(vd)[[3]]}' ",
"-c1 '{colours$treat[[names(vd)[[1]]]]}' ",
"-c2 '{colours$treat[[names(vd)[[2]]]]}' ",
"-c3 '{colours$treat[[names(vd)[[3]]]]}' ",
"-ht {knitr::opts_chunk$get('fig.height')} ",
"-w {knitr::opts_chunk$get('fig.width')} ",
"-o '{fig_out}'"
)
system2(which_py, args)
} 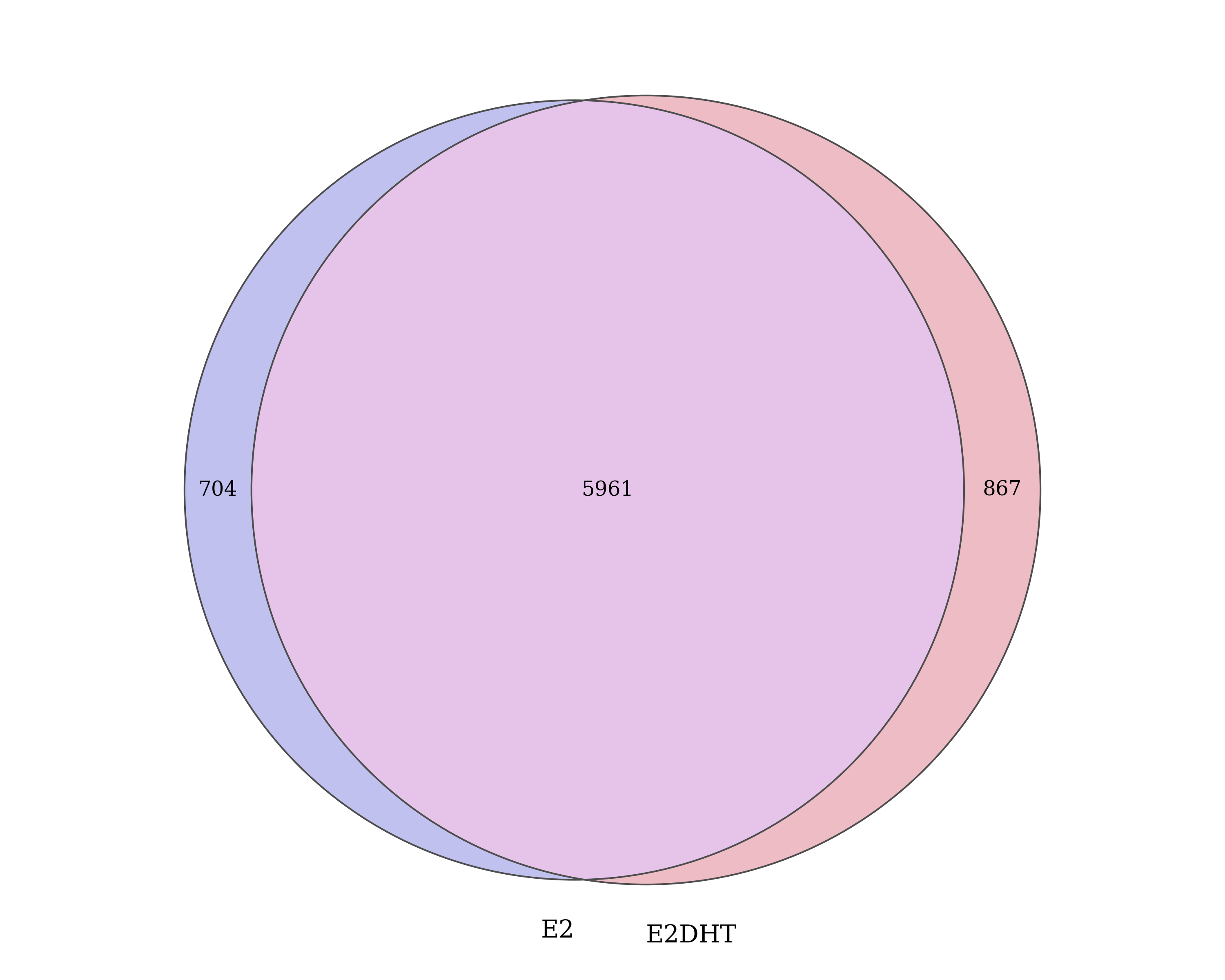
Number of ER union peaks which overlap treatment peaks defined within each condition.
Distance to TSS
a <- union_peaks %>%
join_nearest(
mutate(tss, tss = start)
) %>%
mutate(d = start + width/2 - tss) %>%
as_tibble() %>%
ggplot(aes(d / 1e3)) +
geom_density() +
coord_cartesian(xlim = c(-100, 100)) +
labs(
x = "Distance to Nearest TSS (kb)",
y = "Density"
)
b <- union_peaks %>%
join_nearest(
mutate(tss, tss = start)
) %>%
mutate(d = start + width/2 - tss) %>%
as_tibble() %>%
select(d) %>%
arrange(abs(d)) %>%
mutate(
q = seq_along(d) / nrow(.)
) %>%
ggplot(aes(abs(d) / 1e3, q)) +
geom_line() +
geom_vline(
xintercept = max(unlist(extra_params$gene_regions$promoters)) / 1e3,
linetype = 2, colour = "grey40"
) +
coord_cartesian(xlim = c(0, 100)) +
labs(
x = "Distance to Nearest TSS (kb)",
y = "Percentile"
) +
scale_y_continuous(labels = percent, breaks = seq(0, 1, by = 0.2)) +
scale_x_continuous(breaks = seq(0, 100, by = 20))
a + b + plot_annotation(tag_levels = "A")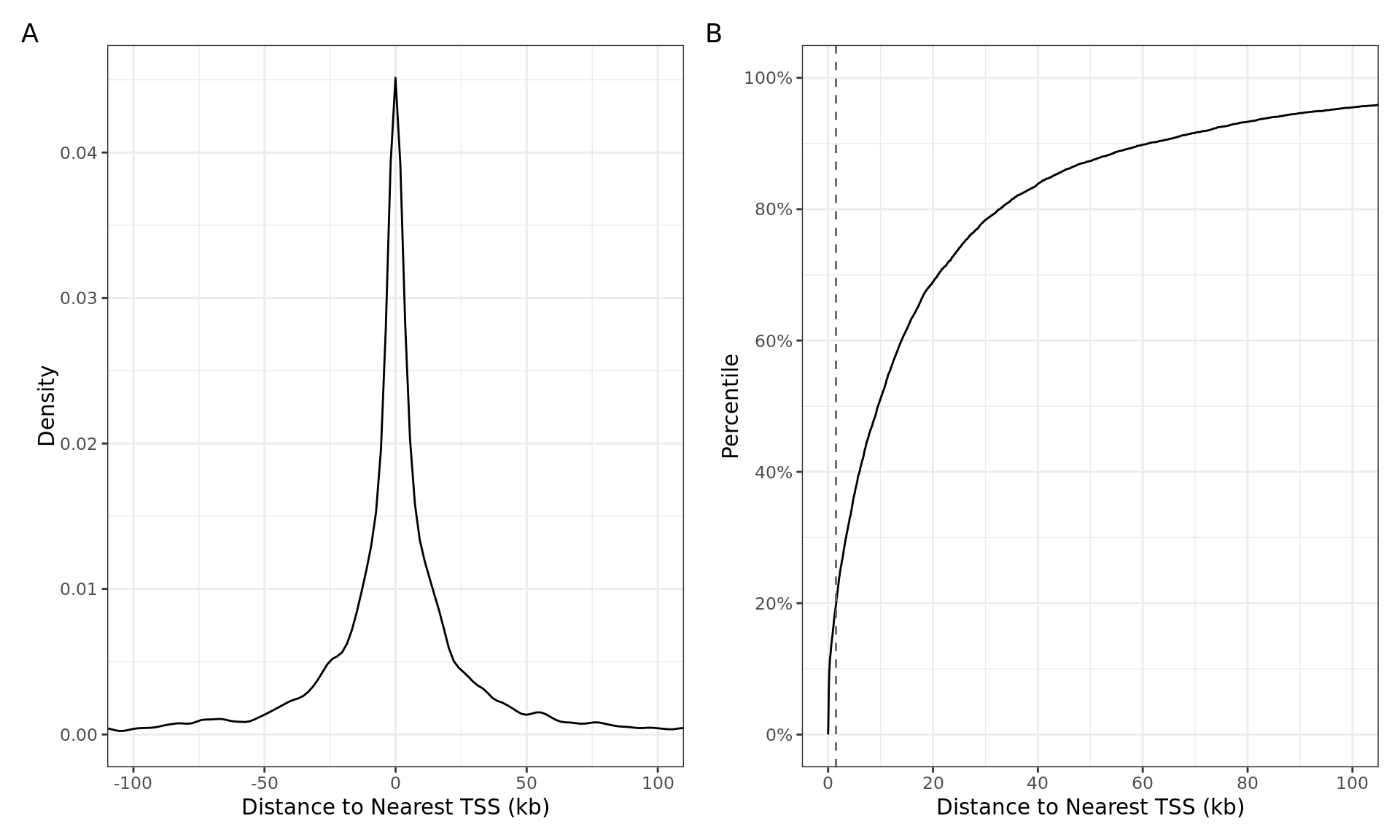
Distances from the centre of the ER union peak to the transcription start-site shown as A) a histogram, and B) as a cumulative distribution. The vertical dashed line indicates the range considered to be a promoter during annotation preparation. 637 of the 7,532 ER union peaks (8.5%) directly overlapped a TSS.
Gene-Centric Regions
union_peaks %>%
plotPie(
fill = "region", total_size = 4,
cat_glue = "{str_wrap(.data[[fill]], 15)}\n{comma(n, 1)}\n({percent(p, 0.1)})",
cat_alpha = 0.9, cat_adj = 0.05, min_p = 0.025
) +
scale_fill_manual(
values = colours$regions %>% setNames(regions[names(.)])
) +
theme(legend.position = "none")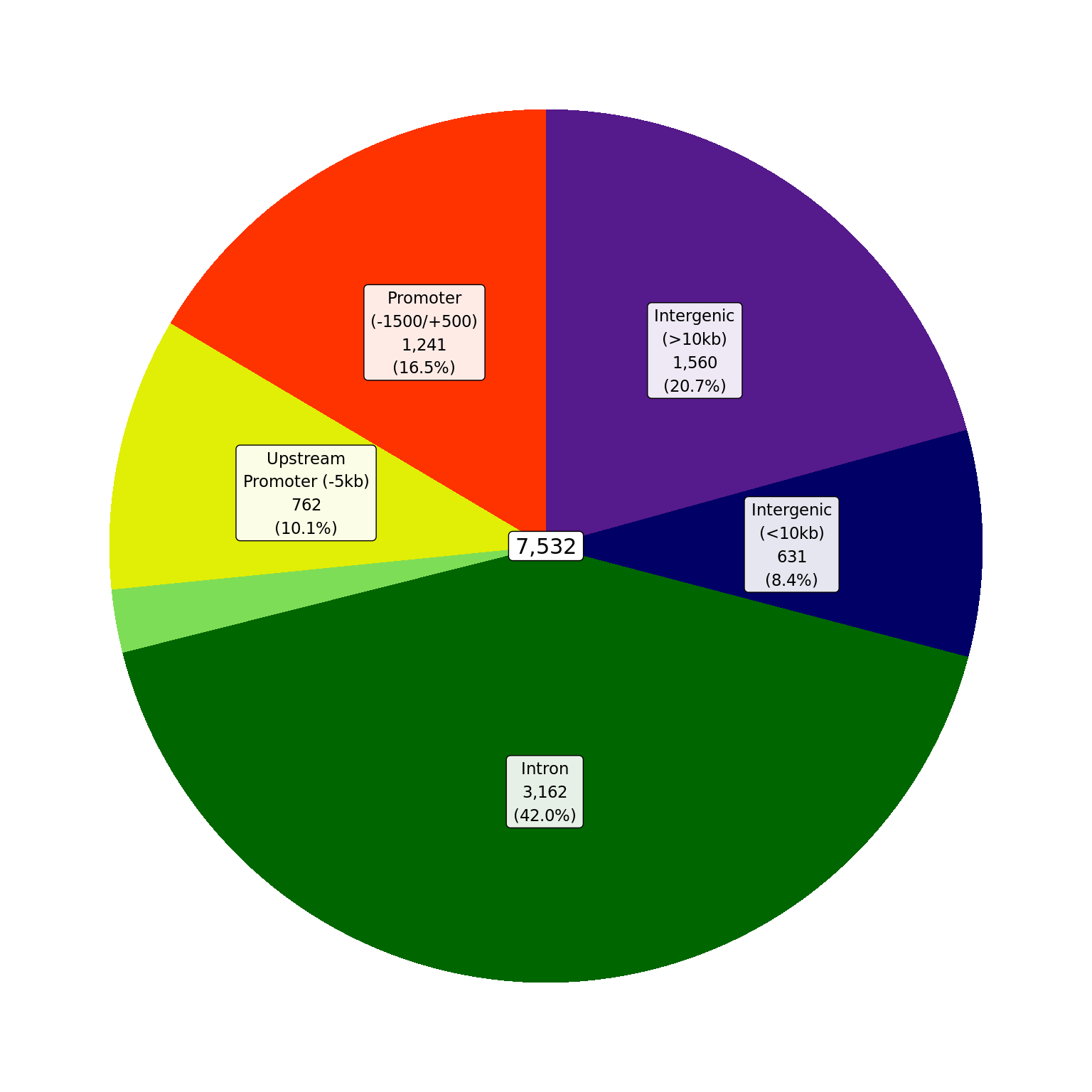
Proportions of ER union peaks which overlap gene-centric features.
External Features
union_peaks %>%
plotPie(
fill = "Feature", total_size = 4,
cat_glue = "{str_wrap(.data[[fill]], 15)}\n{comma(n, 1)}\n({percent(p, 0.1)})",
cat_alpha = 0.9, cat_adj = 0.05
) +
scale_fill_manual(
values = colours$features %>% setNames(str_sep_to_title(names(.)))
) +
theme(legend.position = "none")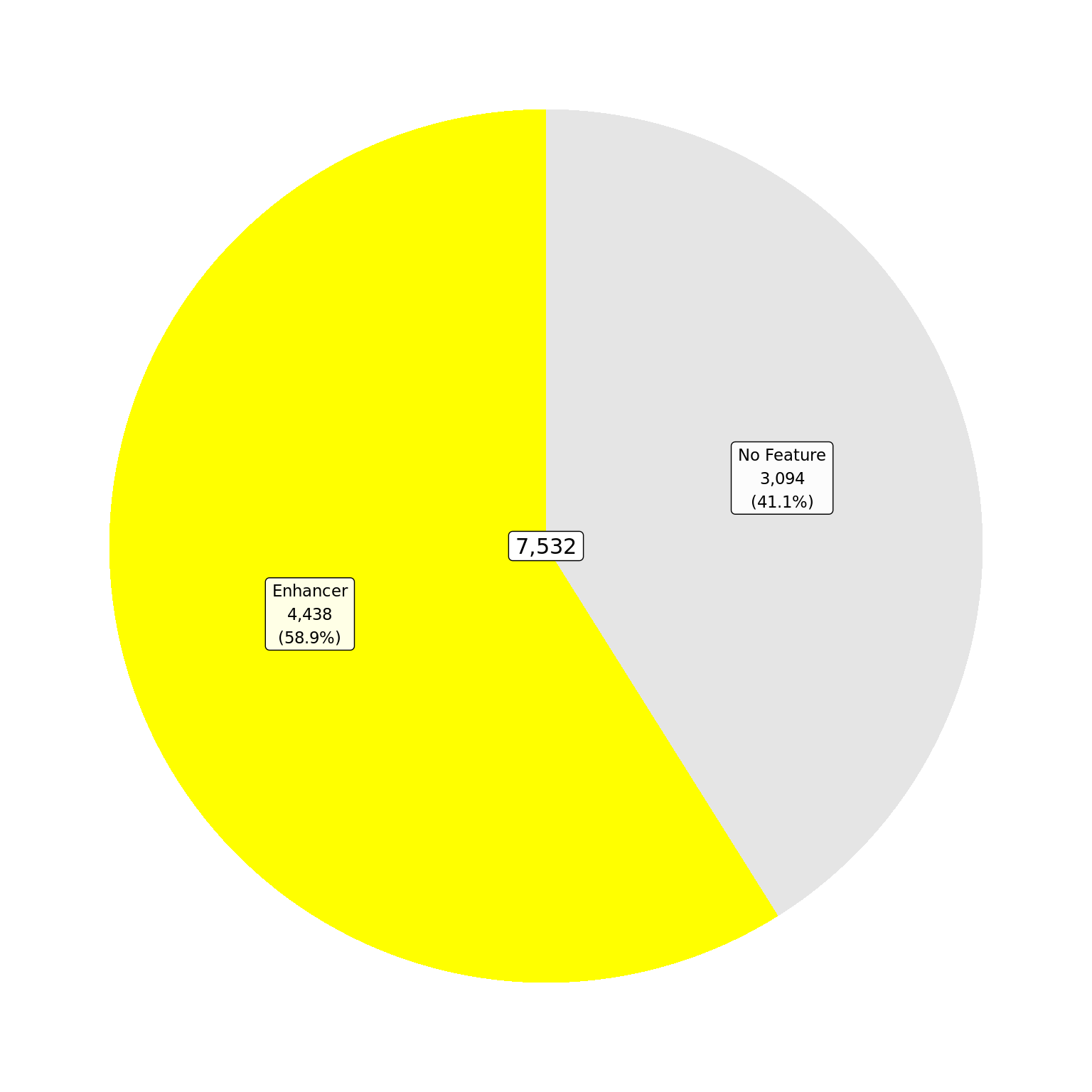
The total number of ER union peaks which overlap external features provided in enhancer_atlas_2.0_zr75.gtf.gz. If peaks map to multiple features, they are assigned to the one with the largest overlap.
External Features And Gene-Centric Regions
union_peaks %>%
plotSplitDonut(
inner = "Feature", outer = "region",
inner_palette = colours$features %>% setNames(str_to_title(names(.))),
outer_palette = colours$regions %>% setNames(regions[names(.)]),
inner_glue = "{str_wrap(.data[[inner]], 15)}\nn = {comma(n, 1)}\n{percent(p,0.1)}",
outer_glue = "{str_wrap(.data[[outer]], 15)}\nn = {comma(n, 1)}\n{percent(p,0.1)}",
min_p = 0.03, outer_alpha = 0.8
)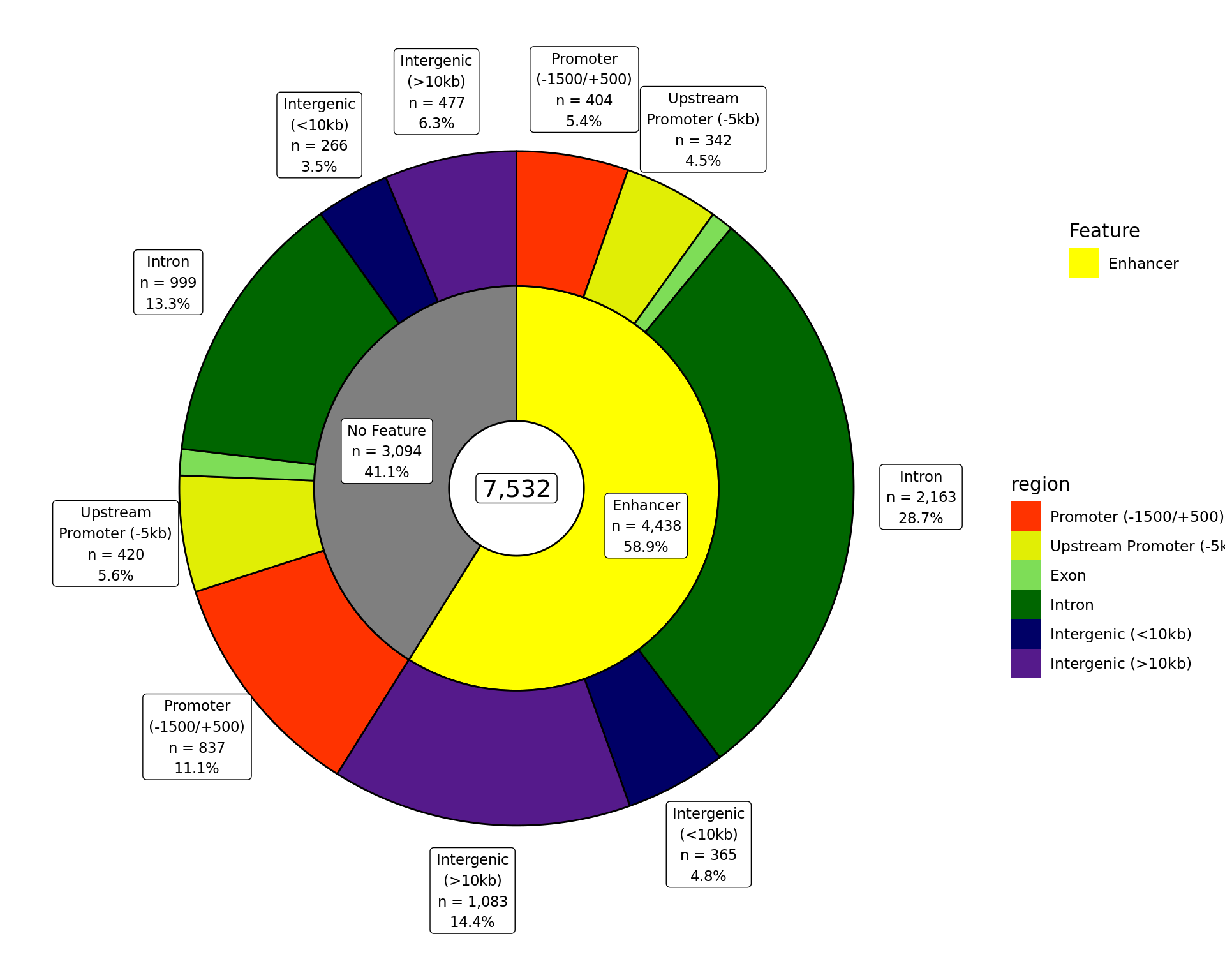
The total number of ER union peaks overlapping external features and gene-centric regions. If a peak overlaps multiple features or regions, it is assigned to be the one with the largest overlap. Any peaks which don’t overlap a feature have been excluded.
Highly Ranked Peaks
grl_to_plot <- vector("list", length(treat_levels) + 1) %>%
setNames(c("union", treat_levels))
grl_to_plot$union <- union_peaks %>%
as_tibble() %>%
arrange(desc(score)) %>%
dplyr::slice(1) %>%
colToRanges("range", seqinfo = sq)
grl_to_plot[treat_levels] <- treat_levels %>%
lapply(
function(x) {
if (length(treatment_peaks[[x]]) == 0) return(NULL)
tbl <- treatment_peaks[[x]] %>%
filter(keep) %>%
filter_by_non_overlaps(
unlist(treatment_peaks[setdiff(treat_levels, x)])
) %>%
filter_by_non_overlaps(grl_to_plot$union) %>%
as_tibble()
if (nrow(tbl) == 0) return(NULL)
tbl %>%
arrange(desc(score)) %>%
dplyr::slice(1) %>%
colToRanges("range", seqinfo = sq)
}
)
grl_to_plot <- grl_to_plot %>%
.[vapply(., length, integer(1)) > 0] %>%
lapply(setNames, NULL) %>%
GRangesList() %>%
unlist() %>%
distinctMC(.keep_all = TRUE) %>%
splitAsList(names(.)) %>%
endoapply(function(x) x[1,]) %>%
.[intersect(c("union", treat_levels), names(.))]## The coverage
bwfl <- list2(
"{target}" := macs2_path %>%
file.path(target, glue("{target}_{treat_levels}_merged_treat_pileup.bw")) %>%
BigWigFileList() %>%
setNames(treat_levels)
)
## The features track
feat_gr <- gene_regions %>%
lapply(granges) %>%
GRangesList()
feature_colours <- colours$regions
if (has_features) {
feat_gr <- list(Regions = feat_gr)
feat_gr$Features <- splitAsList(external_features, external_features$feature)
feature_colours <- list(
Regions = unlist(colours$regions),
Features = unlist(colours$features)
)
}
## The genes track defaults to transcript models
hfgc_genes <- read_rds(
here::here("output", "annotations", "trans_models.rds")
)
gene_col <- "grey"
## If RNA-Seq data is present, the genes track switches to Up/Down etc
if (nrow(rnaseq)) {
rna_lfc_col <- colnames(rnaseq)[str_detect(str_to_lower(colnames(rnaseq)), "logfc")][1]
rna_fdr_col <- colnames(rnaseq)[str_detect(str_to_lower(colnames(rnaseq)), "fdr|adjp")][1]
if (!is.na(rna_lfc_col) & !is.na(rna_fdr_col)) {
hfgc_genes <- hfgc_genes %>%
mutate(
status = case_when(
!gene %in% rnaseq$gene_id ~ "Undetected",
gene %in% dplyr::filter(
rnaseq, !!sym(rna_lfc_col) > 0, !!sym(rna_fdr_col) < fdr_alpha
)$gene_id ~ "Up",
gene %in% dplyr::filter(
rnaseq, !!sym(rna_lfc_col) < 0, !!sym(rna_fdr_col) < fdr_alpha
)$gene_id ~ "Down",
gene %in% dplyr::filter(
rnaseq, !!sym(rna_fdr_col) >= fdr_alpha
)$gene_id ~ "Unchanged"
)
) %>%
splitAsList(.$status) %>%
lapply(select, -status) %>%
GRangesList()
gene_col <- colours$direction %>%
setNames(str_to_title(names(.)))
}
}
## External Coverage (Optional)
if (!is.null(config$external$coverage)) {
ext_cov_path <- config$external$coverage %>%
lapply(unlist) %>%
lapply(function(x) setNames(here::here(x), names(x)))
bwfl <- c(
bwfl[target],
config$external$coverage %>%
lapply(
function(x) {
BigWigFileList(here::here(unlist(x))) %>%
setNames(names(x))
}
)
)
}
line_col <- lapply(bwfl, function(x) colours$treat[names(x)])
y_lim <- bwfl %>%
lapply(
function(x) {
gr <- unlist(grl_to_plot) %>%
resize(width = 30 * width(.), fix = 'center')
x %>%
lapply(import.bw, which = gr) %>%
lapply(function(rng) c(0, max(rng$score))) %>%
unlist() %>%
range()
}
)Coverage for a small set of highly ranked peaks are shown below.
These are the most highly ranked Union Peak (by score) and
any treatment peaks which are unique to each treatment group, after
excluding the most highly ranked union peak.
htmltools::tagList(
mclapply(
seq_along(grl_to_plot),
function(x) {
nm <- names(grl_to_plot)[[x]]
## Export the figure
fig_out <- file.path(
fig_path,
nm %>%
str_replace_all(" ", "_") %>%
paste0("_topranked.", fig_type)
)
fig_fun(
filename = fig_out,
width = knitr::opts_current$get("fig.width"),
height = knitr::opts_current$get("fig.height")
)
## Automatically collapse Transcripts if more than 10
ct <- FALSE
gh <- 1
if (length(subsetByOverlaps(gtf_transcript, grl_to_plot[[x]])) > 20) {
ct <- "meta"
gh <- 0.5
}
## Generate the plot
plotHFGC(
grl_to_plot[[x]],
features = feat_gr, featcol = feature_colours, featsize = 1 + has_features,
genes = hfgc_genes, genecol = gene_col,
coverage = bwfl, linecol = line_col,
cytobands = bands_df,
rotation.title = 90,
zoom = 30,
ylim = y_lim,
collapseTranscripts = ct, genesize = gh,
col.title = "black", background.title = "white",
showAxis = FALSE
)
dev.off()
## Define the caption
gr <- join_nearest(grl_to_plot[[x]], gtf_gene, distance = TRUE)
d <- gr$distance
gn <- gr$gene_name
peak_desc <- ifelse(
nm == "union",
"union peak by combined score across all treatments.",
paste(
"treatment-peak unique to the merged", nm, "samples."
)
)
cp <- htmltools::tags$em(
glue(
"The most highly ranked {peak_desc} ",
ifelse(
d == 0,
paste('The peak directly overlaps', gn),
paste0("The nearest gene was ", gn, ", ", round(d/1e3, 1), "kb away")
),
ifelse(
(nrow(rnaseq) > 0) & (gn %in% gtf_gene$gene_name),
glue(" which was detected in the RNA-Seq data."),
glue(".")
),
"
Y-axes for each coverage track are set to the most highly ranked peak
across all conditions.
"
)
)
## Create html tags
fig_link <- str_extract(fig_out, "assets.+")
htmltools::div(
htmltools::div(
id = nm %>%
str_replace_all(" ", "-") %>%
str_to_lower() %>%
paste0("-topranked"),
class = "section level3",
htmltools::h3(
ifelse(
nm == "union",
"Union Peaks",
paste("Treatment Peaks:", nm)
)
),
htmltools::div(
class = "figure", style = "text-align: center",
htmltools::img(src = fig_link, width = 960),
htmltools::p(
class = "caption", htmltools::tags$em(cp)
)
)
)
)
},
mc.cores = min(length(grl_to_plot), threads)
)
) Union Peaks
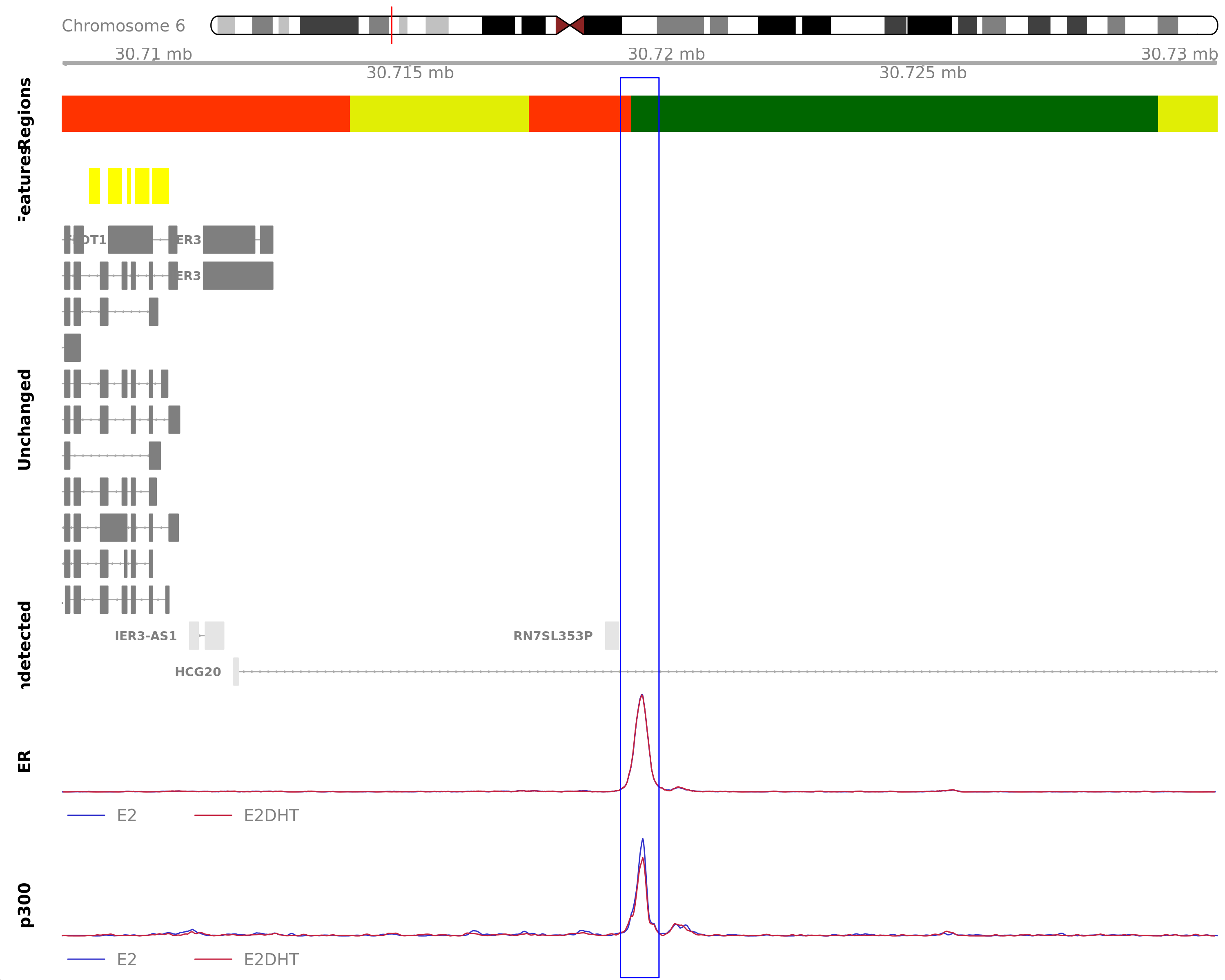
The most highly ranked union peak by combined score across all treatments. The nearest gene was IER3, 6.8kb away which was detected in the RNA-Seq data. Y-axes for each coverage track are set to the most highly ranked peak across all conditions.
Treatment Peaks: E2
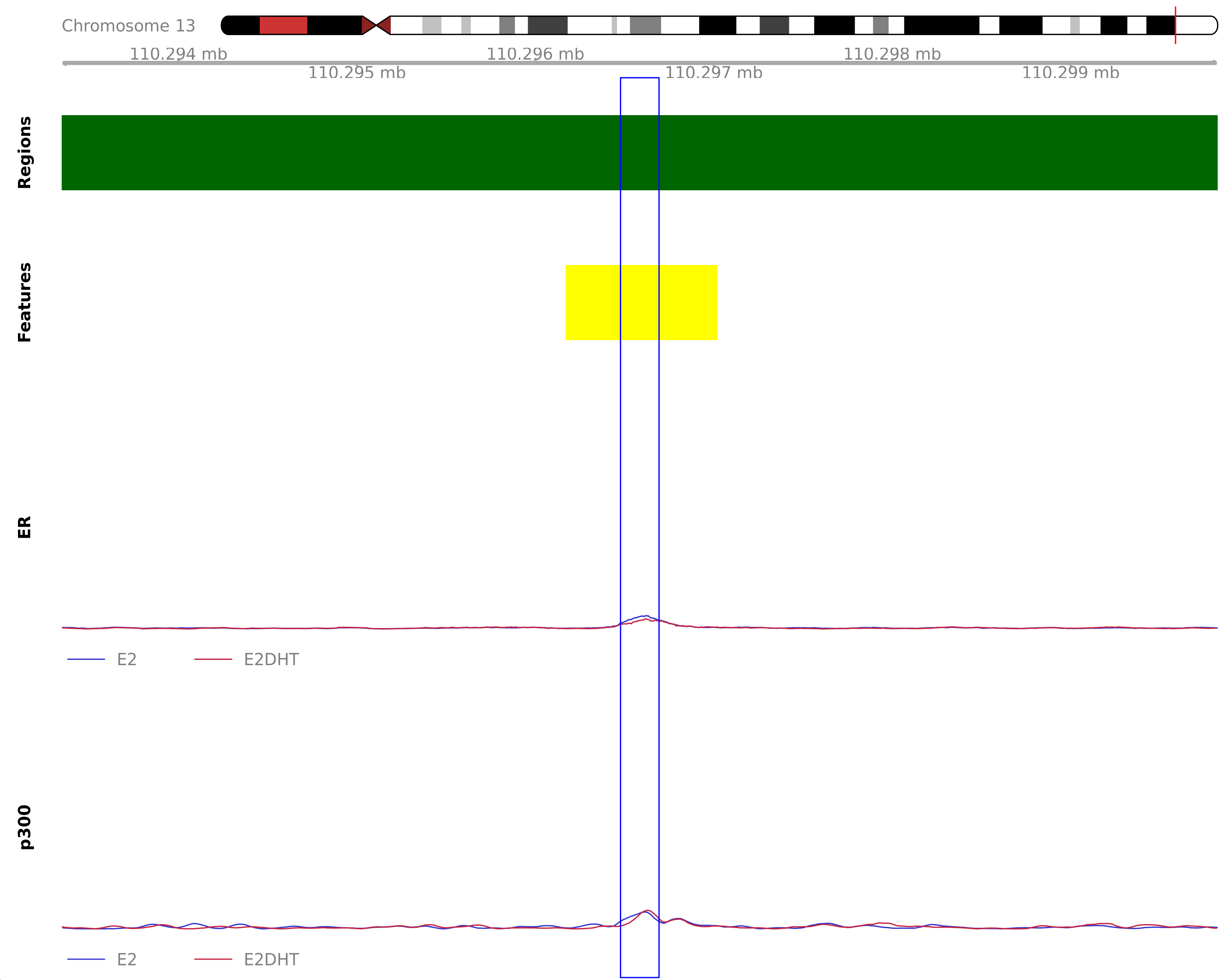
The most highly ranked treatment-peak unique to the merged E2 samples. The nearest gene was IRS2, 108.3kb away which was detected in the RNA-Seq data. Y-axes for each coverage track are set to the most highly ranked peak across all conditions.
Treatment Peaks: E2DHT
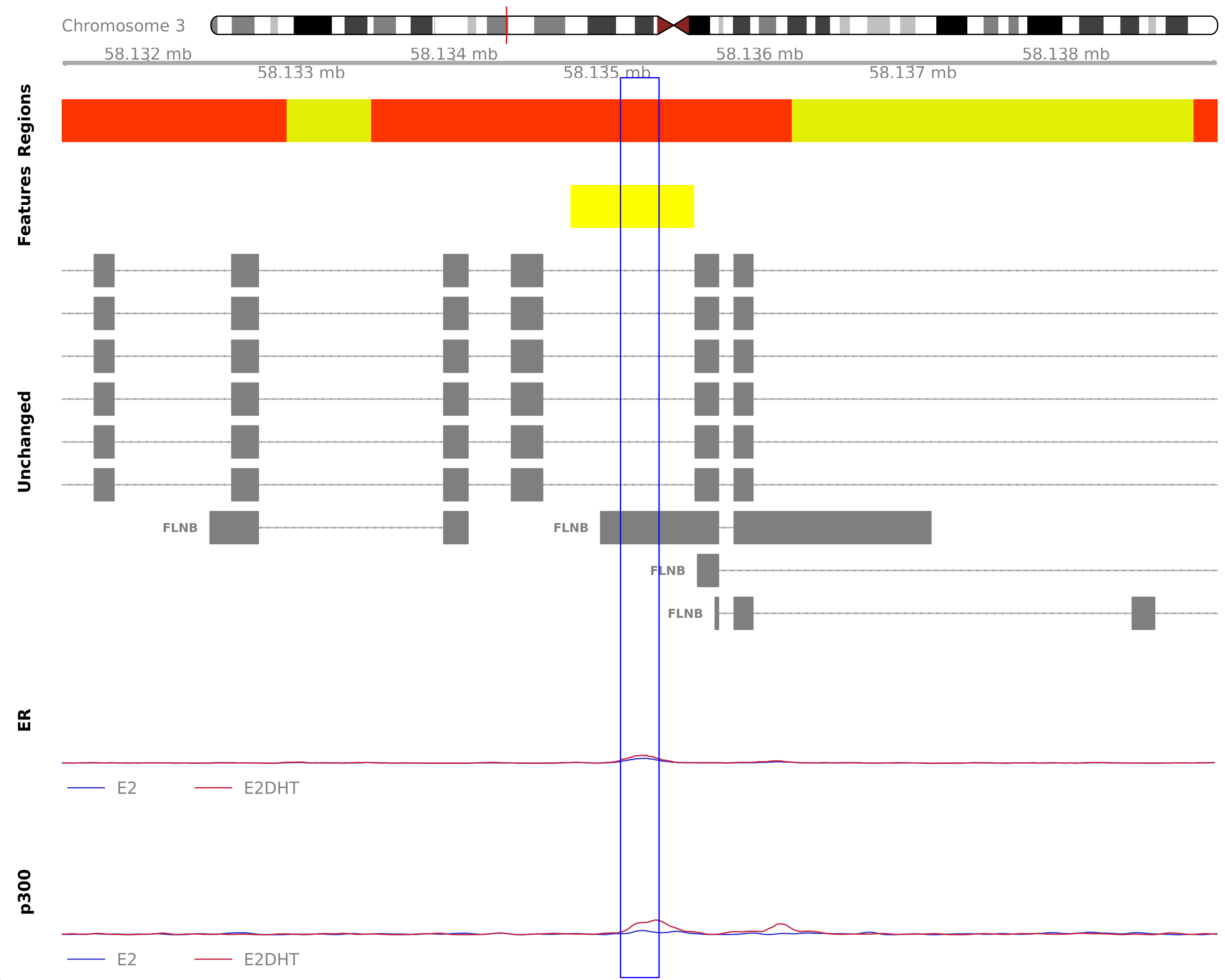
The most highly ranked treatment-peak unique to the merged E2DHT samples. The peak directly overlaps FLNB which was detected in the RNA-Seq data. Y-axes for each coverage track are set to the most highly ranked peak across all conditions.
Data Export
all_out <- list(
union_peaks_bed = file.path(
macs2_path, target, glue("{target}_union_peaks.bed")
),
treatment_peaks_rds = file.path(
macs2_path, target, glue("{target}_treatment_peaks.rds")),
renv = file.path(
here::here("output/envs"), glue("{target}_macs2_summary.RData")
)
) %>%
c(
sapply(
names(treatment_peaks),
function(x) {
file.path(
macs2_path, target, glue("{target}_{x}_treatment_peaks.bed")
)
},
simplify = FALSE
) %>%
setNames(
glue("{target}_{names(.)}_treatment_peaks_bed")
)
)
export(union_peaks, all_out$union_peaks)
treatment_peaks %>%
lapply(subset, keep) %>%
lapply(select, -keep) %>%
GRangesList() %>%
write_rds(all_out$treatment_peaks, compress = "gz")
names(treatment_peaks) %>%
lapply(
function(x) {
id <- glue("{target}_{x}_treatment_peaks_bed")
## Create an empty file
file.create(all_out[[id]])
treatment_peaks[[x]] %>%
subset(keep) %>%
select(score, signalValue, pValue, qValue, peak) %>%
export(all_out[[id]], format = "narrowPeak")
}
)
if (!dir.exists(dirname(all_out$renv))) dir.create(dirname(all_out$renv))
save.image(all_out$renv)During this workflow the following files were exported:
- union_peaks_bed: output/macs2/ER/ER_union_peaks.bed
- treatment_peaks_rds: output/macs2/ER/ER_treatment_peaks.rds
- renv: output/envs/ER_macs2_summary.RData
- ER_E2_treatment_peaks_bed: output/macs2/ER/ER_E2_treatment_peaks.bed
- ER_E2DHT_treatment_peaks_bed: output/macs2/ER/ER_E2DHT_treatment_peaks.bed
References
R version 4.2.3 (2023-03-15)
Platform: x86_64-conda-linux-gnu (64-bit)
locale: LC_CTYPE=en_AU.UTF-8, LC_NUMERIC=C, LC_TIME=en_AU.UTF-8, LC_COLLATE=en_AU.UTF-8, LC_MONETARY=en_AU.UTF-8, LC_MESSAGES=en_AU.UTF-8, LC_PAPER=en_AU.UTF-8, LC_NAME=C, LC_ADDRESS=C, LC_TELEPHONE=C, LC_MEASUREMENT=en_AU.UTF-8 and LC_IDENTIFICATION=C
attached base packages: parallel, grid, stats4, stats, graphics, grDevices, utils, datasets, methods and base
other attached packages: extraChIPs(v.1.5.6), SummarizedExperiment(v.1.28.0), Biobase(v.2.58.0), MatrixGenerics(v.1.10.0), matrixStats(v.1.0.0), ggside(v.0.2.2), Rsamtools(v.2.14.0), Biostrings(v.2.66.0), XVector(v.0.38.0), BiocParallel(v.1.32.5), rlang(v.1.1.1), VennDiagram(v.1.7.3), futile.logger(v.1.4.3), ComplexUpset(v.1.3.3), ngsReports(v.2.0.0), patchwork(v.1.1.2), yaml(v.2.3.7), plyranges(v.1.18.0), scales(v.1.2.1), pander(v.0.6.5), glue(v.1.6.2), rtracklayer(v.1.58.0), GenomicRanges(v.1.50.0), GenomeInfoDb(v.1.34.9), IRanges(v.2.32.0), S4Vectors(v.0.36.0), BiocGenerics(v.0.44.0), magrittr(v.2.0.3), lubridate(v.1.9.2), forcats(v.1.0.0), stringr(v.1.5.0), dplyr(v.1.1.2), purrr(v.1.0.1), readr(v.2.1.4), tidyr(v.1.3.0), tibble(v.3.2.1), ggplot2(v.3.4.2) and tidyverse(v.2.0.0)
loaded via a namespace (and not attached): utf8(v.1.2.3), tidyselect(v.1.2.0), RSQLite(v.2.3.1), AnnotationDbi(v.1.60.0), htmlwidgets(v.1.6.2), munsell(v.0.5.0), codetools(v.0.2-19), interp(v.1.1-4), DT(v.0.28), withr(v.2.5.0), colorspace(v.2.1-0), filelock(v.1.0.2), highr(v.0.10), knitr(v.1.43), rstudioapi(v.0.14), labeling(v.0.4.2), GenomeInfoDbData(v.1.2.9), polyclip(v.1.10-4), farver(v.2.1.1), bit64(v.4.0.5), rprojroot(v.2.0.3), vctrs(v.0.6.3), generics(v.0.1.3), lambda.r(v.1.2.4), xfun(v.0.39), biovizBase(v.1.46.0), timechange(v.0.2.0), csaw(v.1.32.0), BiocFileCache(v.2.6.0), R6(v.2.5.1), doParallel(v.1.0.17), clue(v.0.3-64), locfit(v.1.5-9.8), AnnotationFilter(v.1.22.0), bitops(v.1.0-7), cachem(v.1.0.8), DelayedArray(v.0.24.0), vroom(v.1.6.3), BiocIO(v.1.8.0), nnet(v.7.3-19), gtable(v.0.3.3), ensembldb(v.2.22.0), splines(v.4.2.3), GlobalOptions(v.0.1.2), lazyeval(v.0.2.2), dichromat(v.2.0-0.1), broom(v.1.0.5), checkmate(v.2.2.0), GenomicFeatures(v.1.50.2), backports(v.1.4.1), Hmisc(v.5.1-0), EnrichedHeatmap(v.1.27.2), tools(v.4.2.3), jquerylib(v.0.1.4), RColorBrewer(v.1.1-3), ggdendro(v.0.1.23), Rcpp(v.1.0.10), base64enc(v.0.1-3), progress(v.1.2.2), zlibbioc(v.1.44.0), RCurl(v.1.98-1.12), prettyunits(v.1.1.1), rpart(v.4.1.19), deldir(v.1.0-9), GetoptLong(v.1.0.5), zoo(v.1.8-12), ggrepel(v.0.9.3), cluster(v.2.1.4), here(v.1.0.1), data.table(v.1.14.8), futile.options(v.1.0.1), circlize(v.0.4.15), ProtGenerics(v.1.30.0), hms(v.1.1.3), evaluate(v.0.21), XML(v.3.99-0.14), jpeg(v.0.1-10), gridExtra(v.2.3), shape(v.1.4.6), compiler(v.4.2.3), biomaRt(v.2.54.0), crayon(v.1.5.2), htmltools(v.0.5.5), mgcv(v.1.8-42), tzdb(v.0.4.0), Formula(v.1.2-5), DBI(v.1.1.3), tweenr(v.2.0.2), formatR(v.1.14), dbplyr(v.2.3.2), ComplexHeatmap(v.2.14.0), MASS(v.7.3-60), GenomicInteractions(v.1.32.0), rappdirs(v.0.3.3), Matrix(v.1.5-4.1), cli(v.3.6.1), Gviz(v.1.42.0), metapod(v.1.6.0), igraph(v.1.4.3), pkgconfig(v.2.0.3), GenomicAlignments(v.1.34.0), foreign(v.0.8-84), plotly(v.4.10.2), xml2(v.1.3.4), InteractionSet(v.1.26.0), foreach(v.1.5.2), bslib(v.0.5.0), VariantAnnotation(v.1.44.0), digest(v.0.6.31), rmarkdown(v.2.23), htmlTable(v.2.4.1), edgeR(v.3.40.0), restfulr(v.0.0.15), curl(v.5.0.1), rjson(v.0.2.21), nlme(v.3.1-162), lifecycle(v.1.0.3), jsonlite(v.1.8.7), viridisLite(v.0.4.2), limma(v.3.54.0), BSgenome(v.1.66.3), fansi(v.1.0.4), pillar(v.1.9.0), lattice(v.0.21-8), KEGGREST(v.1.38.0), fastmap(v.1.1.1), httr(v.1.4.6), png(v.0.1-8), iterators(v.1.0.14), bit(v.4.0.5), ggforce(v.0.4.1), stringi(v.1.7.12), sass(v.0.4.6), blob(v.1.2.4), latticeExtra(v.0.6-30) and memoise(v.2.0.1)