Plot the Per Sequence Quality Scores for a set of FASTQC reports
plotSeqQuals(
x,
usePlotly = FALSE,
labels,
pattern = ".(fast|fq|bam).*",
pwfCols,
...
)
# S4 method for class 'ANY'
plotSeqQuals(
x,
usePlotly = FALSE,
labels,
pattern = ".(fast|fq|bam).*",
pwfCols,
...
)
# S4 method for class 'character'
plotSeqQuals(
x,
usePlotly = FALSE,
labels,
pattern = ".(fast|fq|bam).*",
pwfCols,
...
)
# S4 method for class 'FastqcData'
plotSeqQuals(
x,
usePlotly = FALSE,
labels,
pattern = ".(fast|fq|bam).*",
pwfCols,
showPwf = TRUE,
counts = FALSE,
alpha = 0.1,
warn = 30,
fail = 20,
colour = "red",
plotlyLegend = FALSE,
...
)
# S4 method for class 'FastqcDataList'
plotSeqQuals(
x,
usePlotly = FALSE,
labels,
pattern = ".(fast|fq|bam).*",
pwfCols,
counts = FALSE,
alpha = 0.1,
warn = 30,
fail = 20,
showPwf = TRUE,
plotType = c("heatmap", "line"),
dendrogram = FALSE,
cluster = FALSE,
scaleFill = NULL,
heatCols = hcl.colors(100, "inferno"),
heat_w = 8,
scaleColour = NULL,
plotlyLegend = FALSE,
...
)Arguments
- x
Can be a
FastqcData,FastqcDataListor path- usePlotly
logicalDefaultFALSEwill render using ggplot. IfTRUEplot will be rendered with plotly- labels
An optional named vector of labels for the file names. All file names must be present in the names of the vector.
- pattern
Regex to remove from the end of any filenames
- pwfCols
Object of class
PwfCols()containing the colours for PASS/WARN/FAIL- ...
Used to pass various potting parameters to theme. Can also be used to set size and colour for box outlines.
- showPwf
logical(1) Show PASS/WARN/FAIL status
- counts
logical. Plot the counts from each file ifcounts = TRUE, otherwise the frequencies will be plotted- alpha
set alpha for line graph bounds
- warn, fail
The default values for warn and fail are 5 and 10 respectively (i.e. percentages)
- colour
Colour for single line plots
- plotlyLegend
logical(1) Show legend for interactive line plots
- plotType
character. Can only take the valuesplotType = "heatmap"orplotType = "line"- dendrogram
logicalredundant ifclusterisFALSEif bothclusteranddendrogramare specified asTRUEthen the dendrogram will be displayed.- cluster
logicaldefaultFALSE. If set toTRUE, fastqc data will be clustered using hierarchical clustering- scaleFill, scaleColour
ggplot2 scales
- heatCols
Colour palette for the heatmap
- heat_w
Relative width of any heatmap plot components
Value
A standard ggplot2 object, or an interactive plotly object
Details
Plots the distribution of average sequence quality scores across the
set of files. Values can be plotted either as counts (counts = TRUE)
or as frequencies (counts = FALSE).
Any faceting or scale adjustment can be performed after generation of the initial plot, using the standard methods of ggplot2 as desired.
Examples
# Get the files included with the package
packageDir <- system.file("extdata", package = "ngsReports")
fl <- list.files(packageDir, pattern = "fastqc.zip", full.names = TRUE)
# Load the FASTQC data as a FastqcDataList object
fdl <- FastqcDataList(fl)
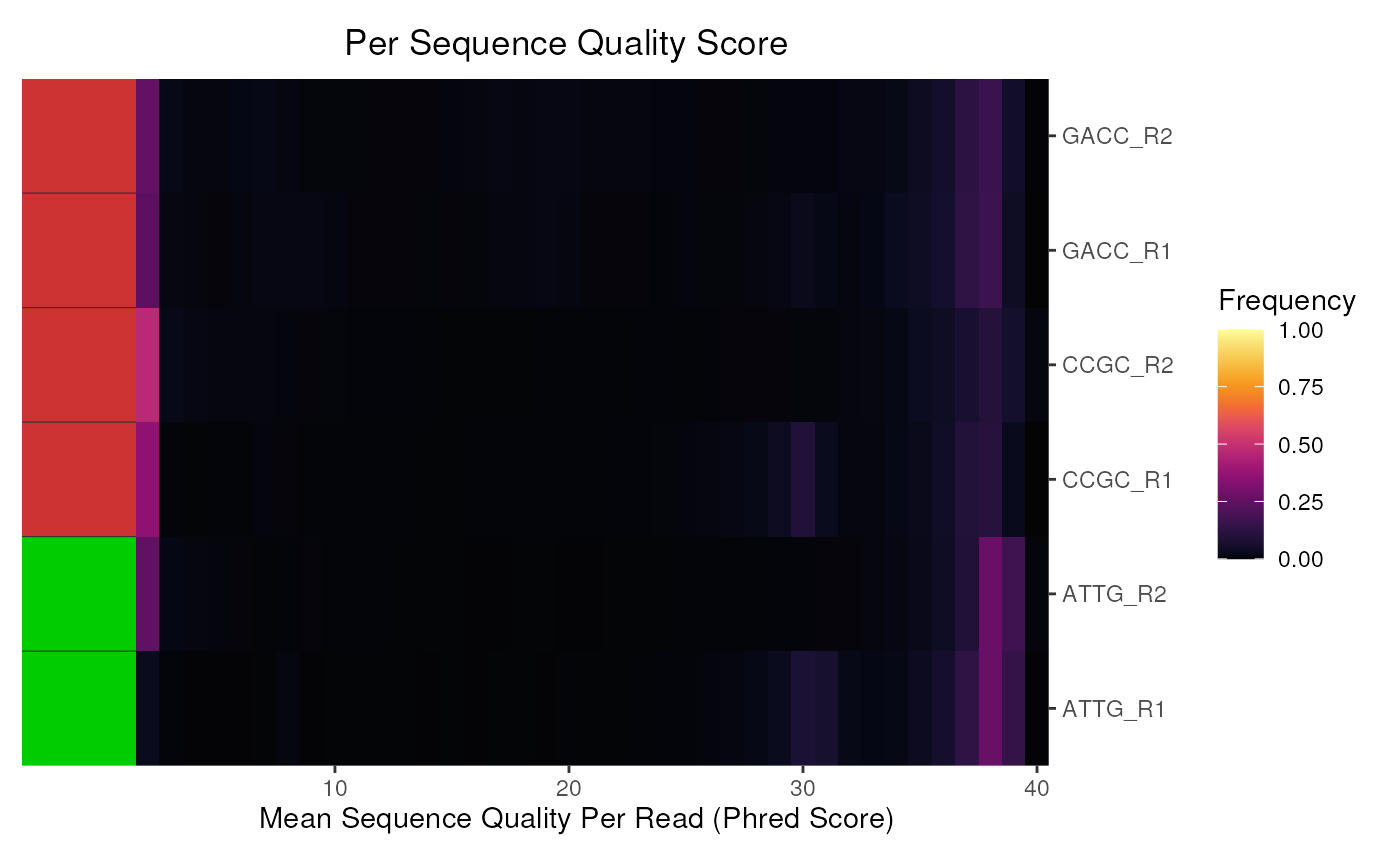
# The default plot
plotSeqQuals(fdl)
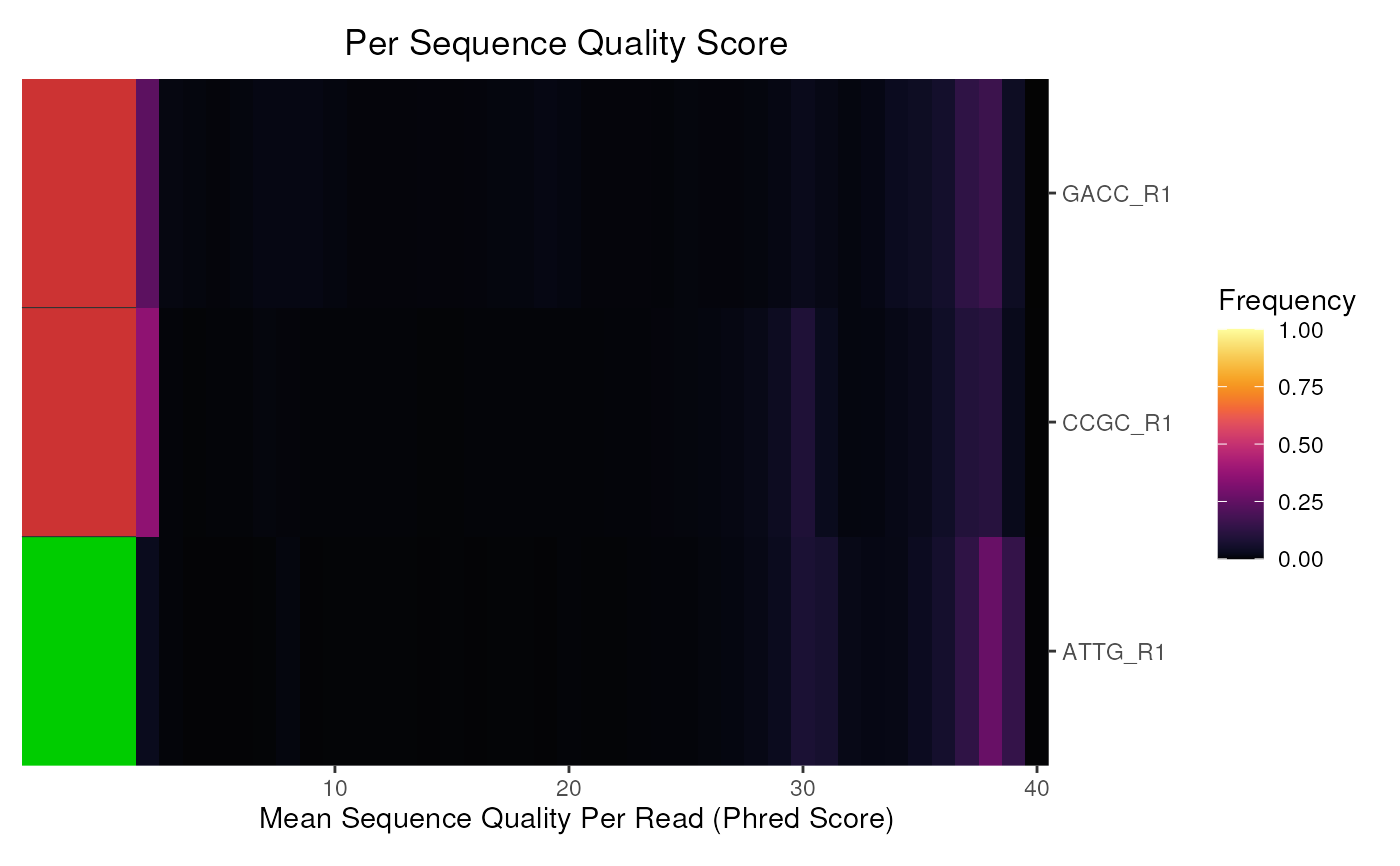
 # Also subset the reads to just the R1 files
r1 <- grepl("R1", fqName(fdl))
plotSeqQuals(fdl[r1])
# Also subset the reads to just the R1 files
r1 <- grepl("R1", fqName(fdl))
plotSeqQuals(fdl[r1])