Plot the Per Base content for a set of FASTQC files.
plotSeqContent(x, usePlotly = FALSE, labels, pattern = ".(fast|fq|bam).*", ...)
# S4 method for class 'ANY'
plotSeqContent(x, usePlotly = FALSE, labels, pattern = ".(fast|fq|bam).*", ...)
# S4 method for class 'FastqcData'
plotSeqContent(
x,
usePlotly = FALSE,
labels,
pattern = ".(fast|fq|bam).*",
bases = c("A", "T", "C", "G"),
scaleColour = NULL,
plotTheme = theme_get(),
plotlyLegend = FALSE,
expand.x = 0.02,
expand.y = c(0, 0.05),
...
)
# S4 method for class 'FastqcDataList'
plotSeqContent(
x,
usePlotly = FALSE,
labels,
pattern = ".(fast|fq|bam).*",
pwfCols,
showPwf = TRUE,
plotType = c("heatmap", "line", "residuals"),
scaleColour = NULL,
plotTheme = theme_get(),
cluster = FALSE,
dendrogram = FALSE,
heat_w = 8,
plotlyLegend = FALSE,
nc = 2,
...
)
# S4 method for class 'FastpData'
plotSeqContent(
x,
usePlotly = FALSE,
labels,
pattern = ".(fast|fq|bam).*",
module = c("Before_filtering", "After_filtering"),
reads = c("read1", "read2"),
readsBy = c("facet", "linetype"),
moduleBy = c("facet", "linetype"),
bases = c("A", "T", "C", "G", "N", "GC"),
scaleColour = NULL,
scaleLine = NULL,
plotlyLegend = FALSE,
plotTheme = theme_get(),
expand.x = 0.02,
expand.y = c(0, 0.05),
...
)
# S4 method for class 'FastpDataList'
plotSeqContent(
x,
usePlotly = FALSE,
labels,
pattern = ".(fast|fq|bam).*",
module = c("Before_filtering", "After_filtering"),
moduleBy = c("facet", "linetype"),
reads = c("read1", "read2"),
readsBy = c("facet", "linetype"),
bases = c("A", "T", "C", "G", "N", "GC"),
showPwf = FALSE,
pwfCols,
warn = 10,
fail = 20,
plotType = c("heatmap", "line", "residuals"),
plotlyLegend = FALSE,
scaleColour = NULL,
scaleLine = NULL,
plotTheme = theme_get(),
cluster = FALSE,
dendrogram = FALSE,
heat_w = 8,
expand.x = c(0.01),
expand.y = c(0, 0.05),
nc = 2,
...
)Arguments
- x
Can be a
FastqcData,FastqcDataListor file paths- usePlotly
logical. Generate an interactive plot using plotly- labels
An optional named vector of labels for the file names. All file names must be present in the names of the vector.
- pattern
Regex to remove from the end of any filenames
- ...
Used to pass additional attributes to plotting geoms
- bases
Which bases to draw on the plot. Also becomes the default plotting order by setting these as factor levels
- scaleColour
Discrete colour scale as a ggplot ScaleDiscrete object If not provided, will default to scale_colour_manual
- plotTheme
theme object to be applied. Note that all plots will have theme_bw theme applied by default, as well as any additional themes supplied here
- plotlyLegend
logical(1) Show legends for interactive plots. Ignored for heatmaps
- expand.x, expand.y
Passed to expansion in the x- and y-axis scales respectively
- pwfCols
Object of class
PwfCols()to give colours for pass, warning, and fail values in plot- showPwf
Show PASS/WARN/FAIL categories as would be defined in a FastQC report
- plotType
character. Type of plot to generate. Must be "line", "heatmap" or "residuals"- cluster
logicaldefaultFALSE. If set toTRUE, fastqc data will be clustered using hierarchical clustering- dendrogram
logicalredundant ifclusterisFALSEif bothclusteranddendrogramare specified asTRUEthen the dendrogram will be displayed.- heat_w
Relative width of any heatmap plot components
- nc
Specify the number of columns if plotting a FastqcDataList as line plots. Passed to facet_wrap.
- module
Fastp Module to show. Can only be Before/After_filtering
- reads
Which set of reads to show
- readsBy, moduleBy
When plotting both R1 & R2 and both modules, separate by either linetype or linetype
- scaleLine
Discrete scale_linetype object. Only relevant if plotting values by linetype
- warn, fail
Default values for WARN and FAIL based on FastQC reports. Only applied to heatmaps for FastpDataList objects
Value
A ggplot2 object or an interactive plotly object
Details
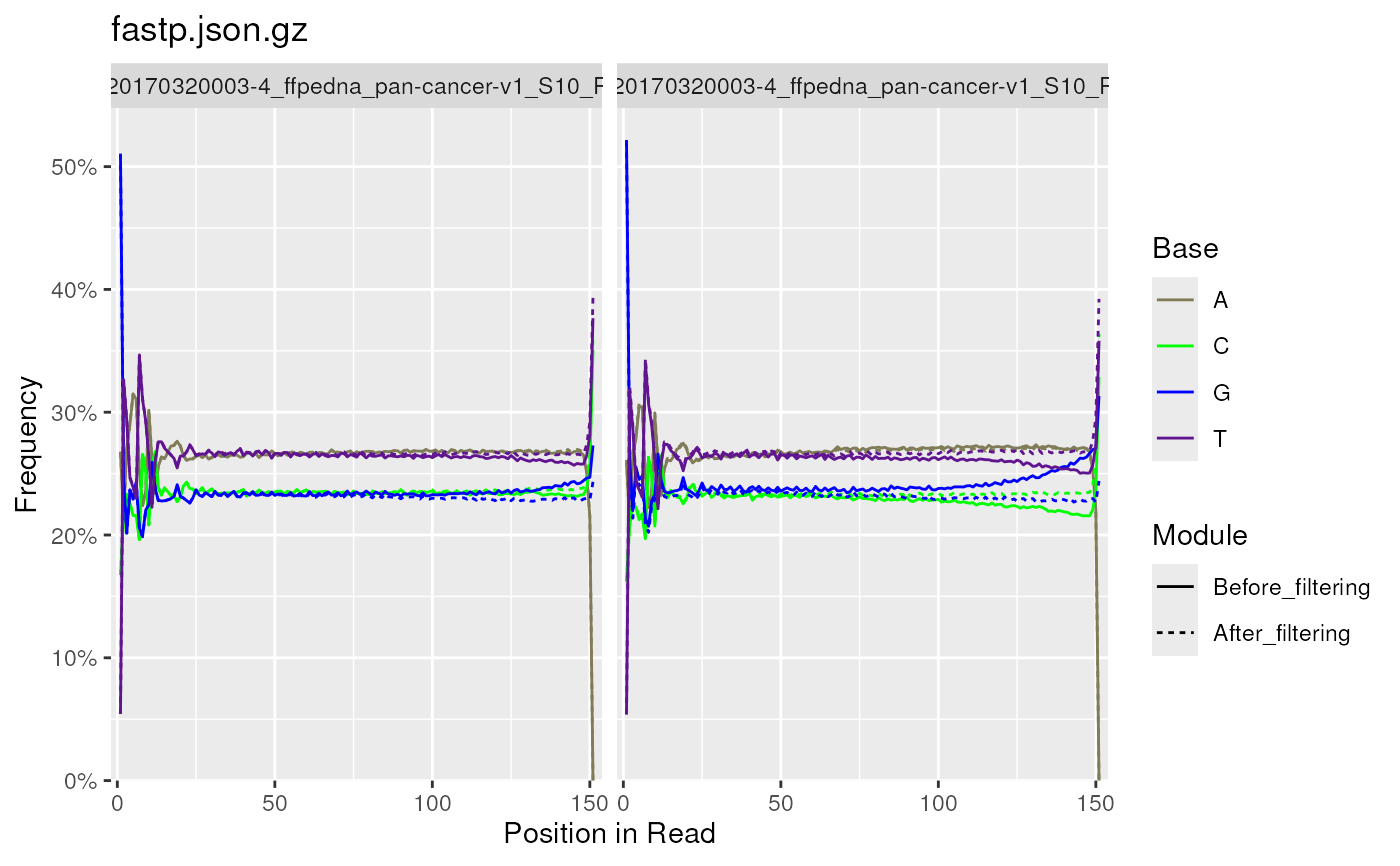
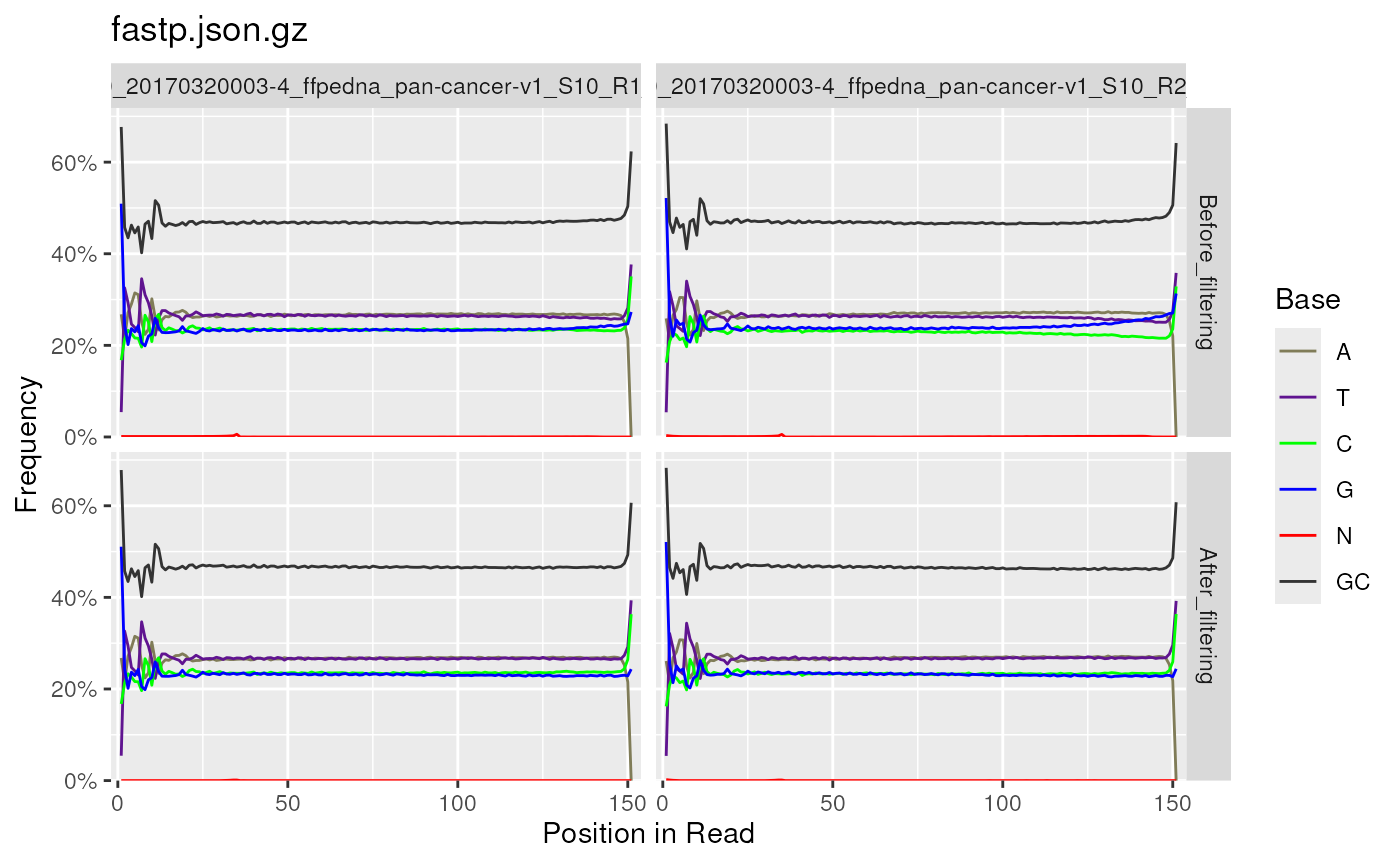
Per base sequence content (%A, %T, %G, %C), is shown as four overlaid
heatmap colours when plotting from multiple reports. The individual line
plots are able to be generated by setting plotType = "line", and the
layout is determined by facet_wrap from ggplot2.
Individual line plots are also generated when plotting from a single
FastqcData object.
If setting usePlotly = TRUE for a large number of reports, the plot
can be slow to render.
An alternative may be to produce a plot of residuals for each base, produced
by taking the position-specific mean for each base.
Examples
# Get the files included with the package
packageDir <- system.file("extdata", package = "ngsReports")
fl <- list.files(packageDir, pattern = "fastqc.zip", full.names = TRUE)
# Load the FASTQC data as a FastqcDataList object
fdl <- FastqcDataList(fl)
# The default plot
plotSeqContent(fdl)
 fp <- FastpData(system.file("extdata/fastp.json.gz", package = "ngsReports"))
plotSeqContent(fp)
fp <- FastpData(system.file("extdata/fastp.json.gz", package = "ngsReports"))
plotSeqContent(fp)
 plotSeqContent(fp, moduleBy = "linetype", bases = c("A", "C", "G", "T"))
plotSeqContent(fp, moduleBy = "linetype", bases = c("A", "C", "G", "T"))