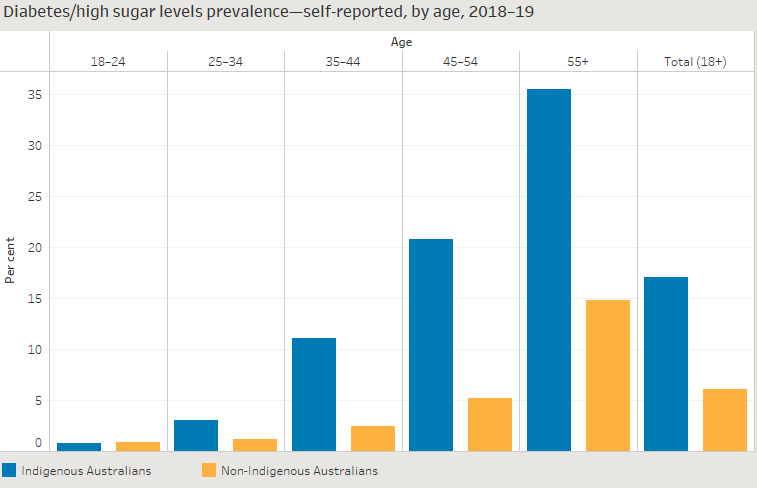
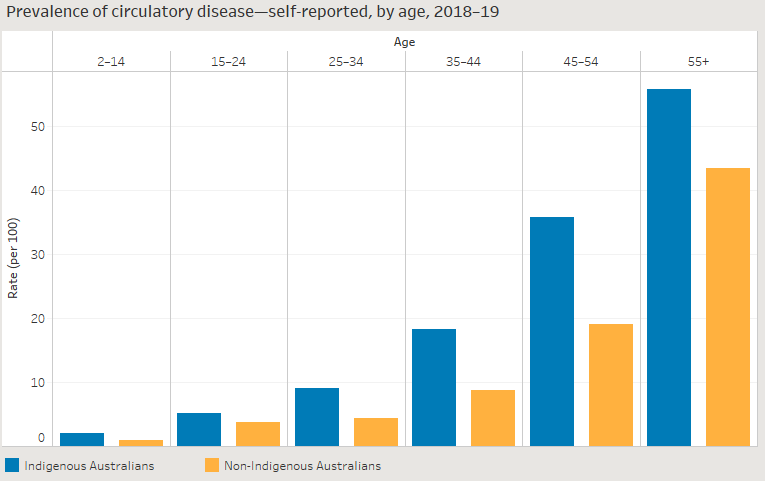
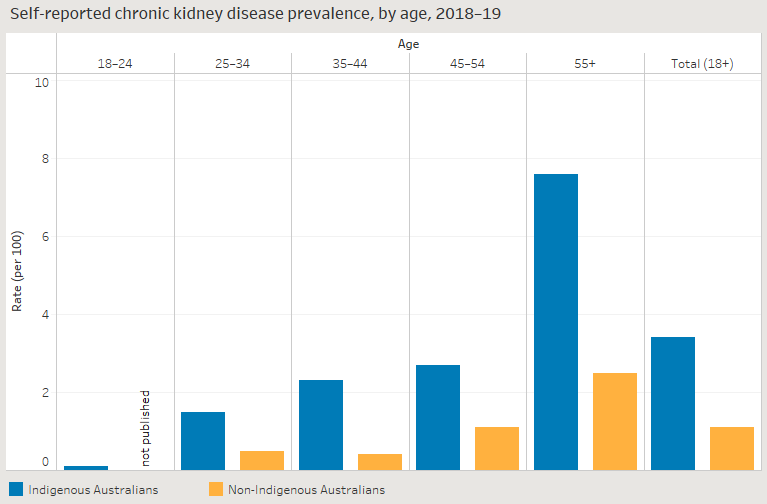
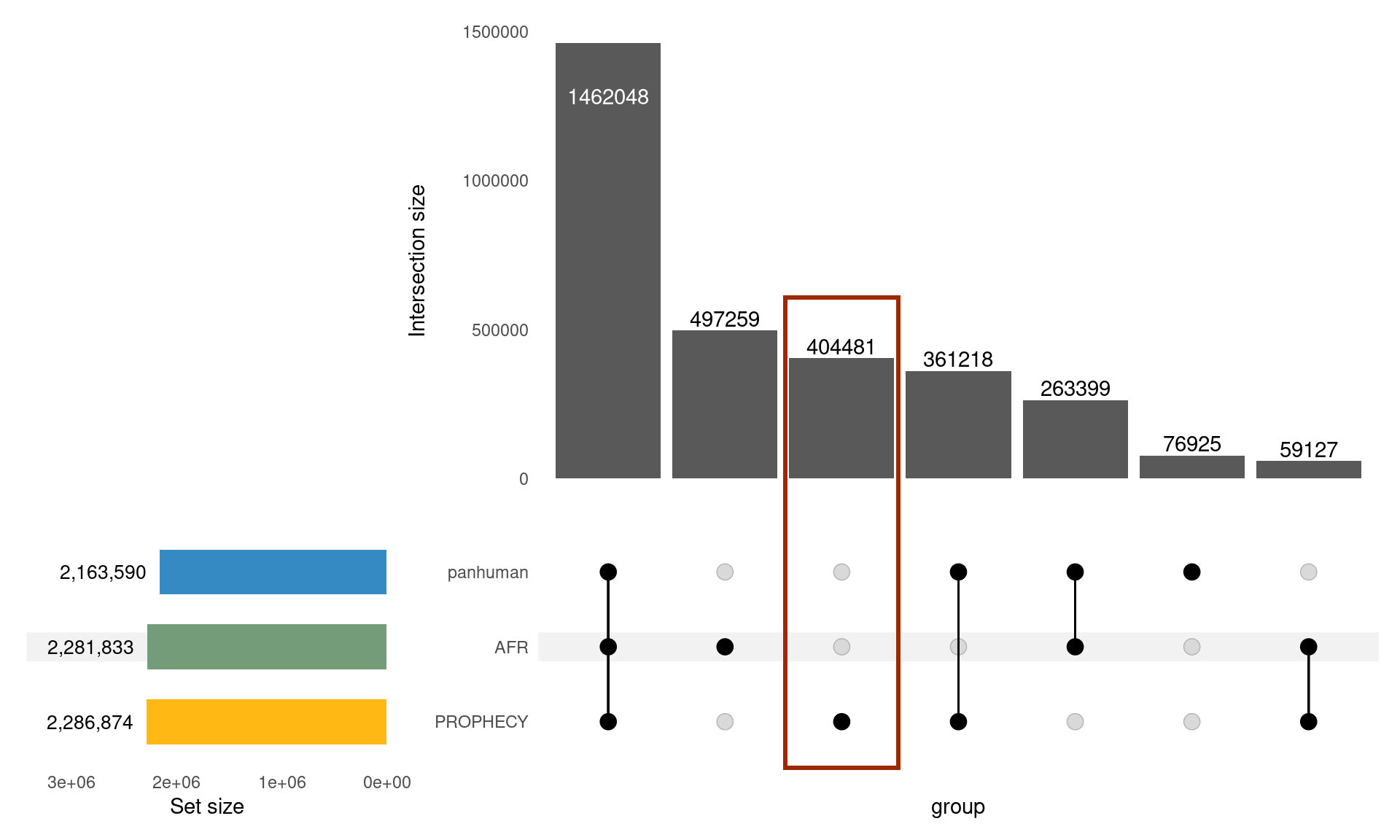
Baldoni, Pedro L, Yunshun Chen, Soroor Hediyeh-Zadeh, Yang Liao, Xueyi Dong, Matthew E Ritchie, Wei Shi, and Gordon K Smyth. 2024. “Dividing Out Quantification Uncertainty Allows Efficient Assessment of Differential Transcript Expression with edgeR.” Nucleic Acids Res. 52 (3): e13.
Easteal, Simon, Ruth M Arkell, Renzo F Balboa, Shayne A Bellingham, Alex D Brown, Tom Calma, Matthew C Cook, et al. 2020. “Equitable Expanded Carrier Screening Needs Indigenous Clinical and Population Genomic Data.” Am. J. Hum. Genet. 107 (2): 175–82.
Kaminow, Benjamin, Sara Ballouz, Jesse Gillis, and Alexander Dobin. 2022. “Pan-Human Consensus Genome Significantly Improves the Accuracy of RNA-seq Analyses.” Genome Res. 32 (4): 738–49.
Srivastava, Avi, Laraib Malik, Hirak Sarkar, Mohsen Zakeri, Fatemeh Almodaresi, Charlotte Soneson, Michael I Love, Carl Kingsford, and Rob Patro. 2020. “Alignment and Mapping Methodology Influence Transcript Abundance Estimation.” Genome Biol. 21 (1): 239.