ALIGN Capacity Building
The Why, What and How of Transcriptomics
November 7, 2023
DNA
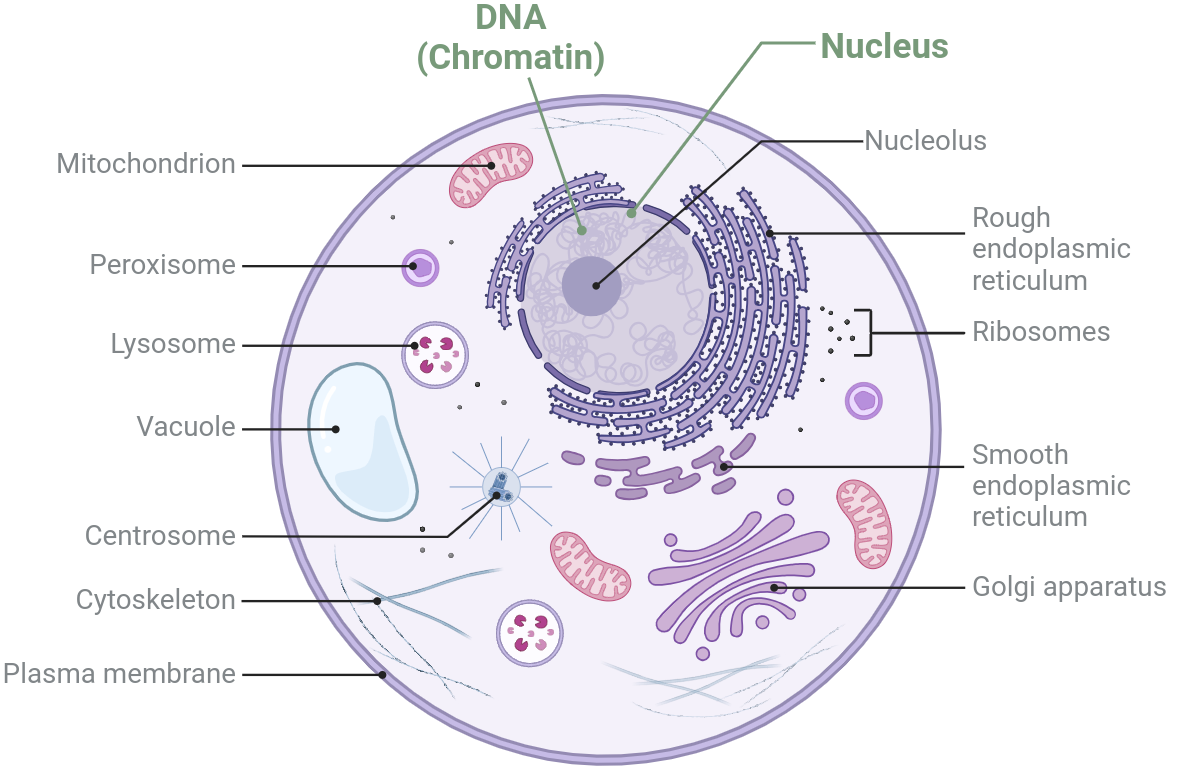
- Most are familiar with DNA and cells
- DNA stays in the nucleus
- Directly inherited from parents
\(\implies\) unique people - The same in every cell (roughly)
- Generally static (until cell division)
- Lots of interaction with other molecules: DNA-methylation, Histones etc.
DNA
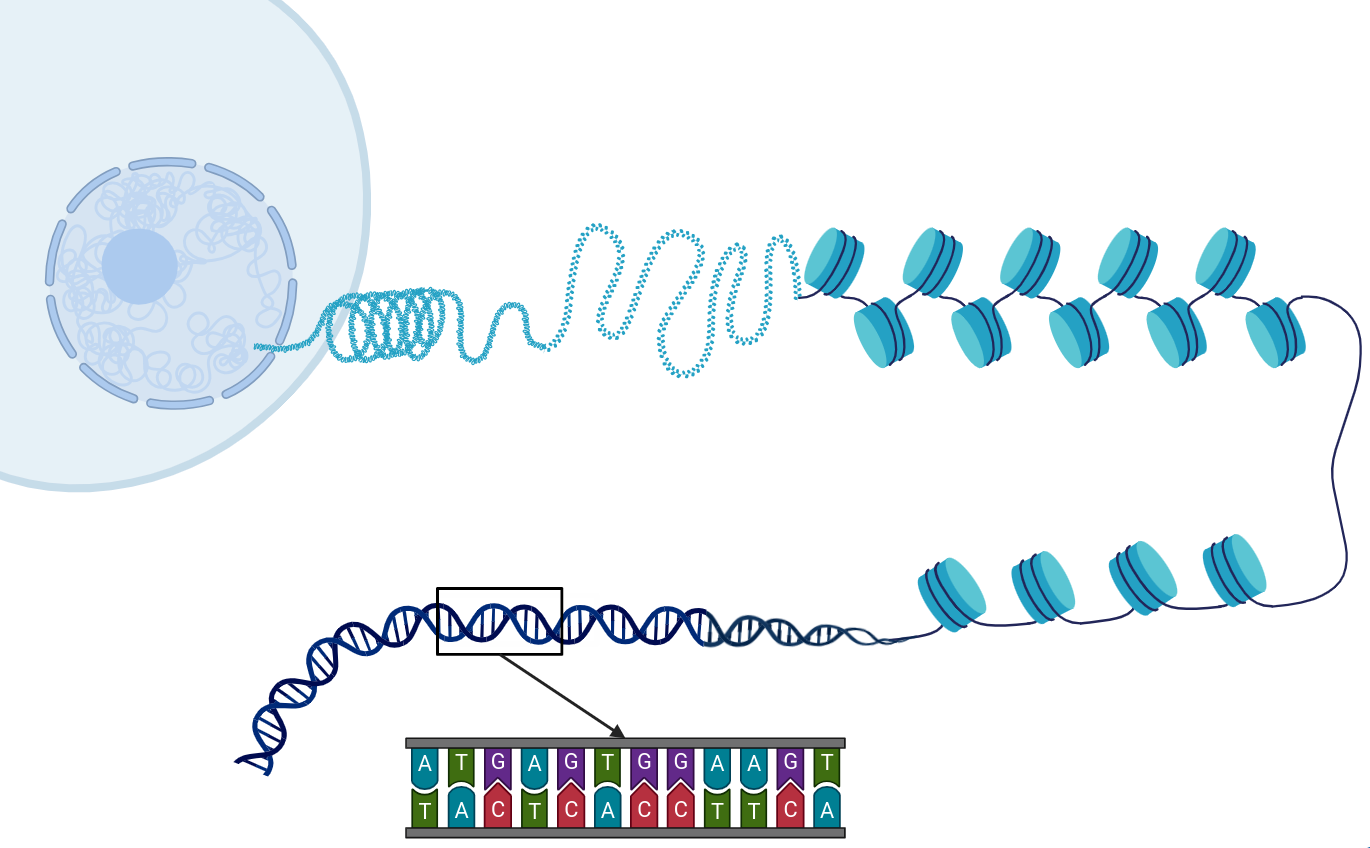
Each chromosome is a string of millions of nucleotides
- 4 nucleotides (or bases): A, C, G, T
- Double-stranded molecule, i.e. double helix
- Adds incredible stability
- Provides error handling mechanism
- Amazing amount of 3D structure & packaging
- Only forms the classic
![]() shape during cell division
shape during cell division
 shape during cell division
shape during cell divisionTranscriptomics
Our classic viewpoint is DNA \(\implies\) mRNA \(\implies\) Protein
Many other types of RNA play key roles
- miRNA bind to and degrade target mRNA
- lncRNA form highly complex structures
\(\implies\)can silence chromosomal regions (e.g. XIST)1
- rRNA are key components of ribosomes
- tRNA interact with ribosomes
\(\implies\) translate mRNA into proteins2

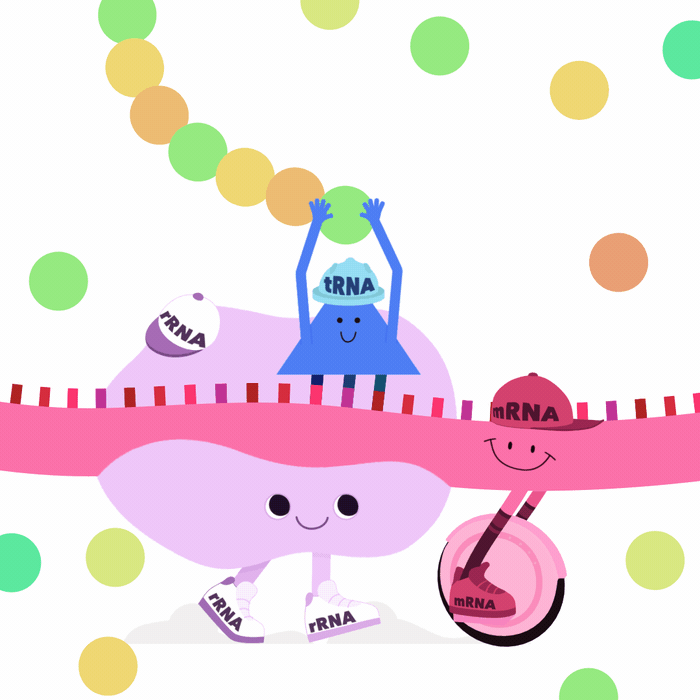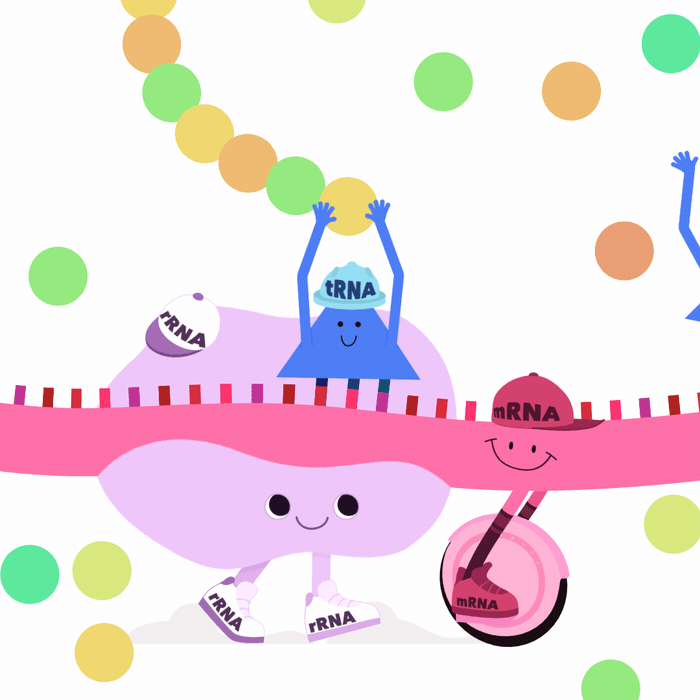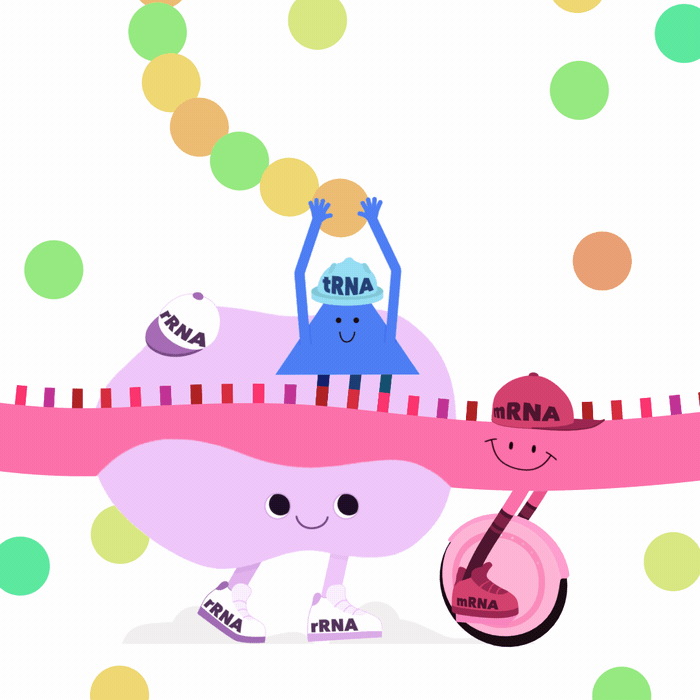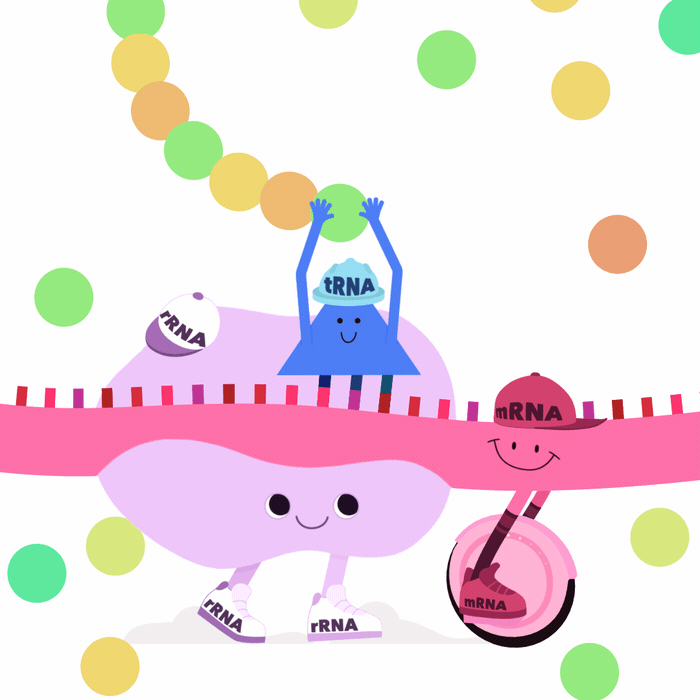
What is an RNA transcript?
- Known transcribed regions are defined as genes
- Transcribed from DNA \(\implies\) start to finish
- From the complete RNA transcript:
- Exons form mature transcript
- Introns are spliced out
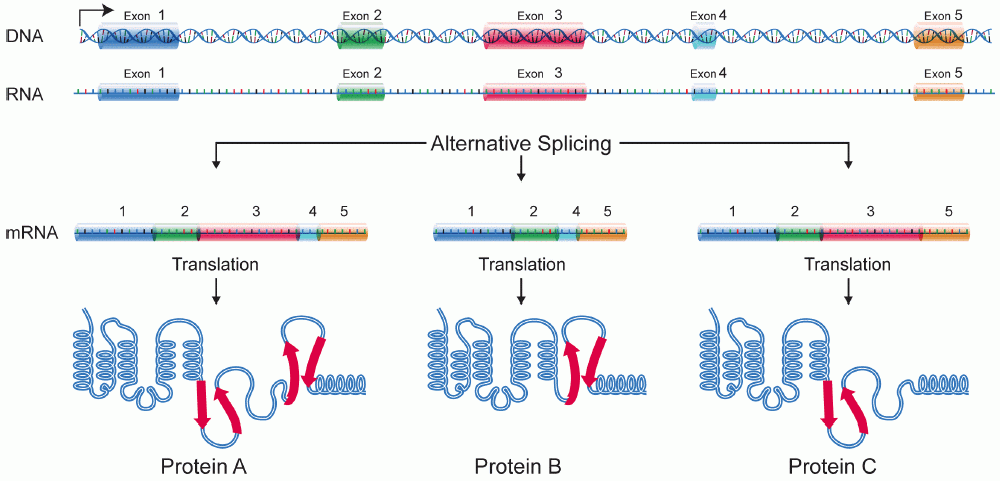
- ~38% of genes (~24,000) are transcribed into multiple transcripts
- Different transcripts \(\implies\) Different proteins (or lncRNA etc.)
Early Gene Expression Approaches
- The first high-throughput platform was Microarrays
- 3’ Arrays (Affymetrix)
- Gene-centric approach
- Could analyse 10-15,000 genes
- Abundance only analysis
- Many statistical tools developed in this context
limmahas been maintained by Gordon Smyth (WEHI) for >20years

Microarrays
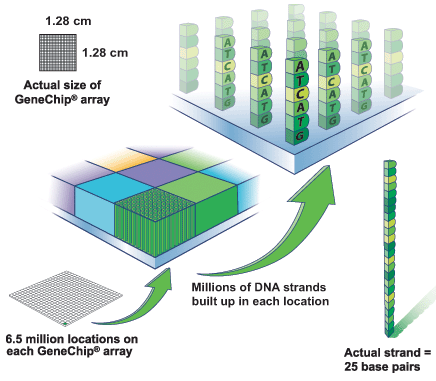
Known sequences at known locations

Labelled cDNA binds complementary target
- We analysed fluorescence NOT sequence data
Bulk RNA Sequencing
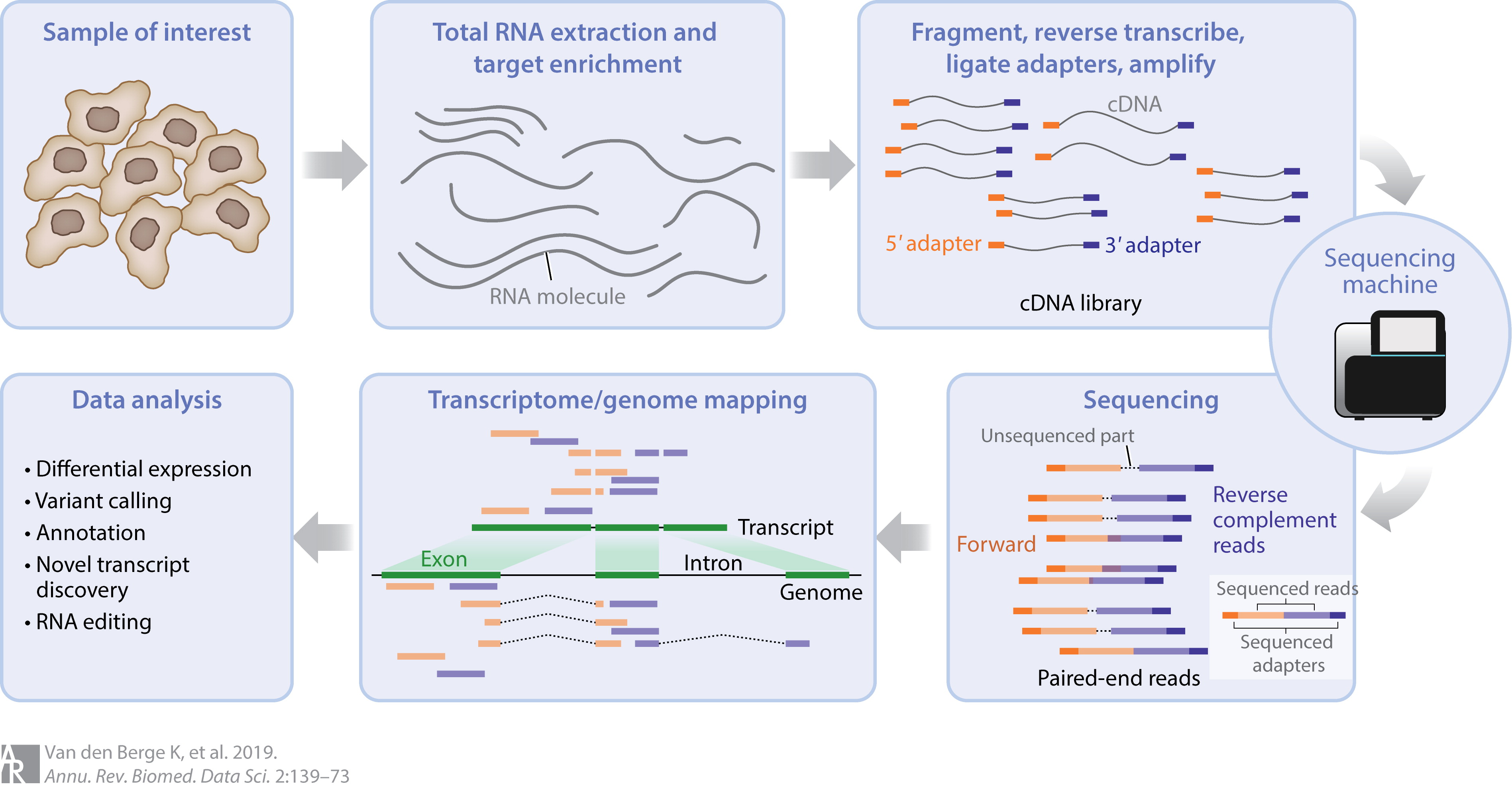
Two Approaches To Alignment
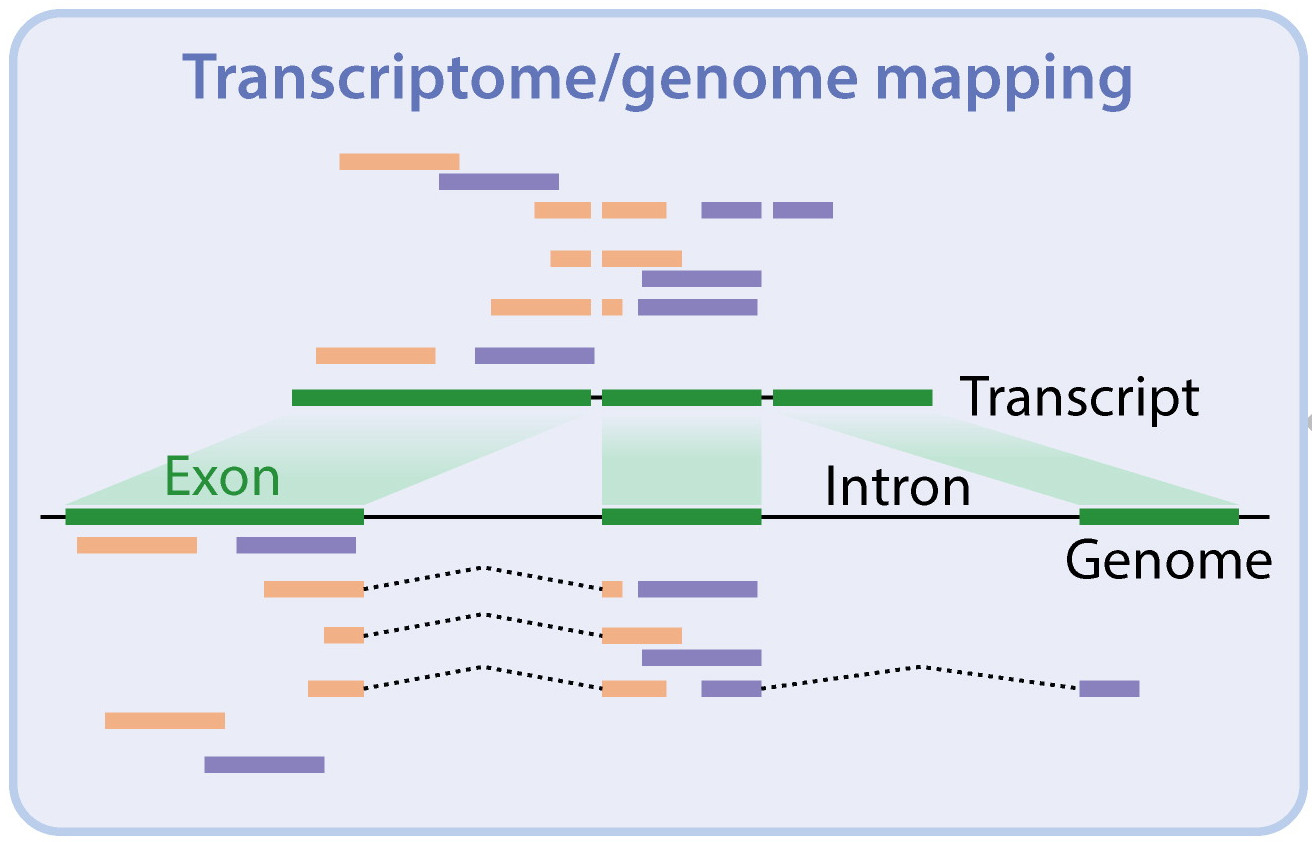
- Alignment to a reference genome
- Most align to one location \(\implies\) can see where ✓
- Brilliant for gene-level counts ✓
- Can call variants (allelic bias) ✓
- Not helpful for transcript-level ✗
- Alignment to a reference transcriptome
- Obtain transcript & gene-level counts ✓
- Many exons shared with multiple transcripts
\(\implies\) can model uncertainty ✓ - Don’t know where each read has aligned ✗
- No variant calling
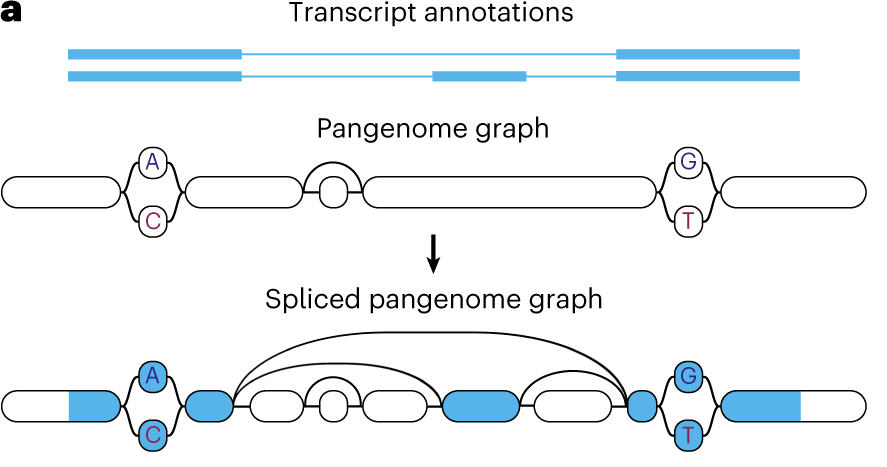
Graph Based Approaches
- Telomere-to-telomere (T2T) assemblies contain no scaffolds
- Just chr1-22, X, Y & MT
- Individual assemblies \(\implies\) haplotype resolved
- Separate out both copies of chr1 etc.
- Can construct a reference graph
- Shared sequences between chromosome pairs are joined
- Variation represented as bubbles
- For >1 individual would be a pangenome graph
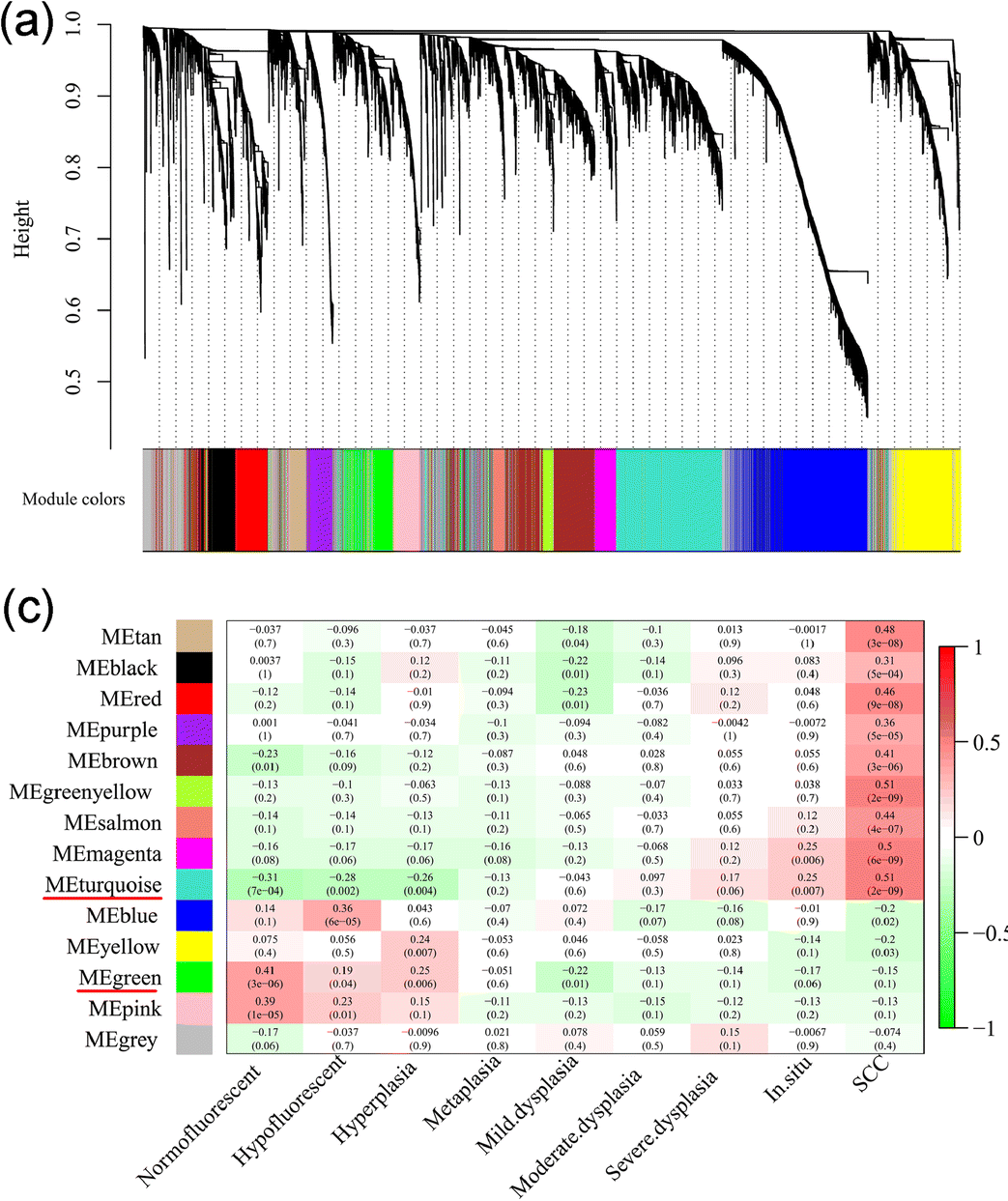
Differential Gene Expression


WGCNA
- Multiple approaches with WGCNA the biggest player
- Form correlation network
- Identify modules within correlation network
- Compare to predictor variables
- Identify underlying biology
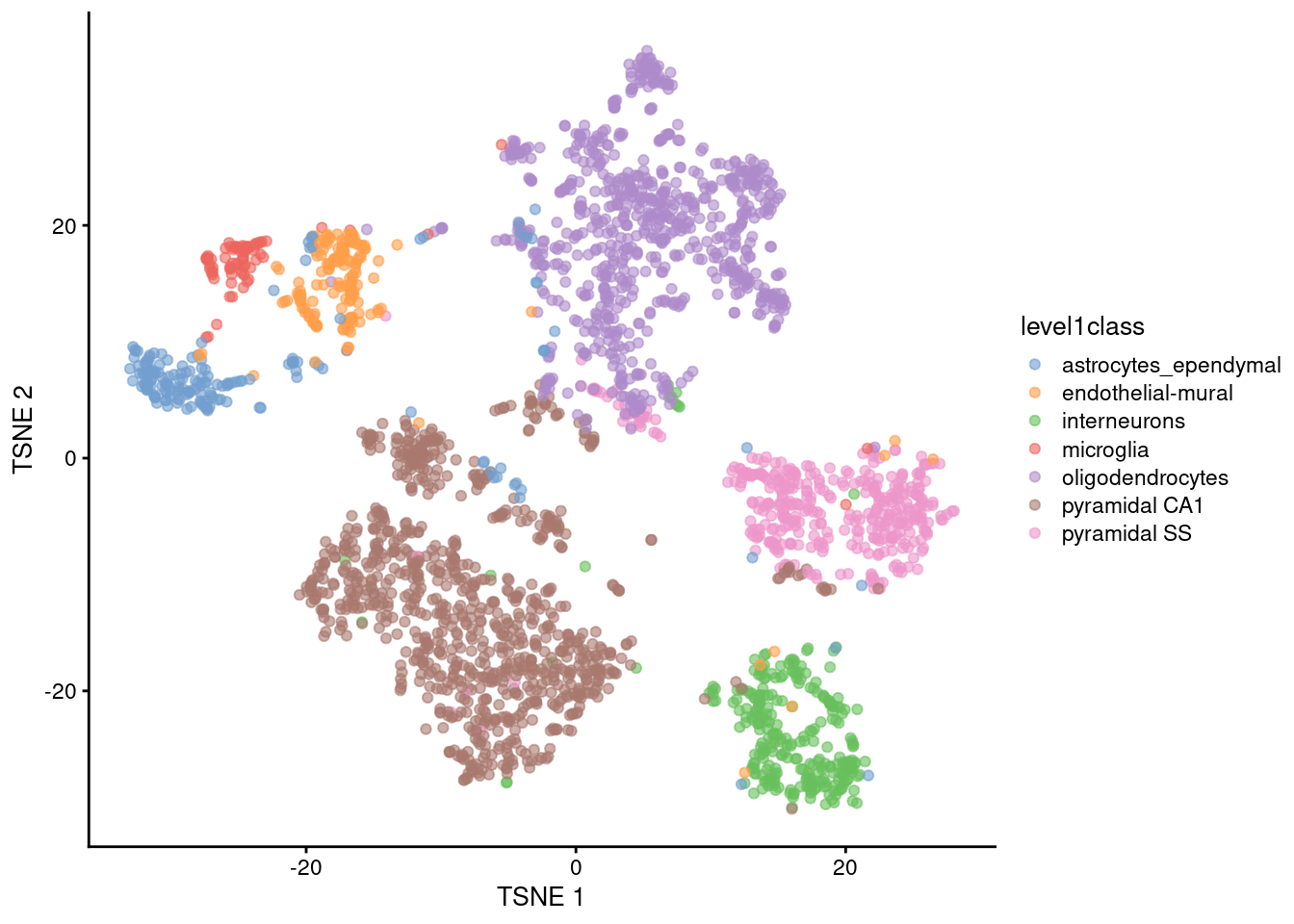
scRNA-Seq
- Retains the connection between transcript and cell-of-origin
- Huge numbers of ‘failure to detect’ expression (i.e. zero counts)
- Uses clustering to identify cell types within a sample
- Pseudo-bulk clusters for DGE analysis
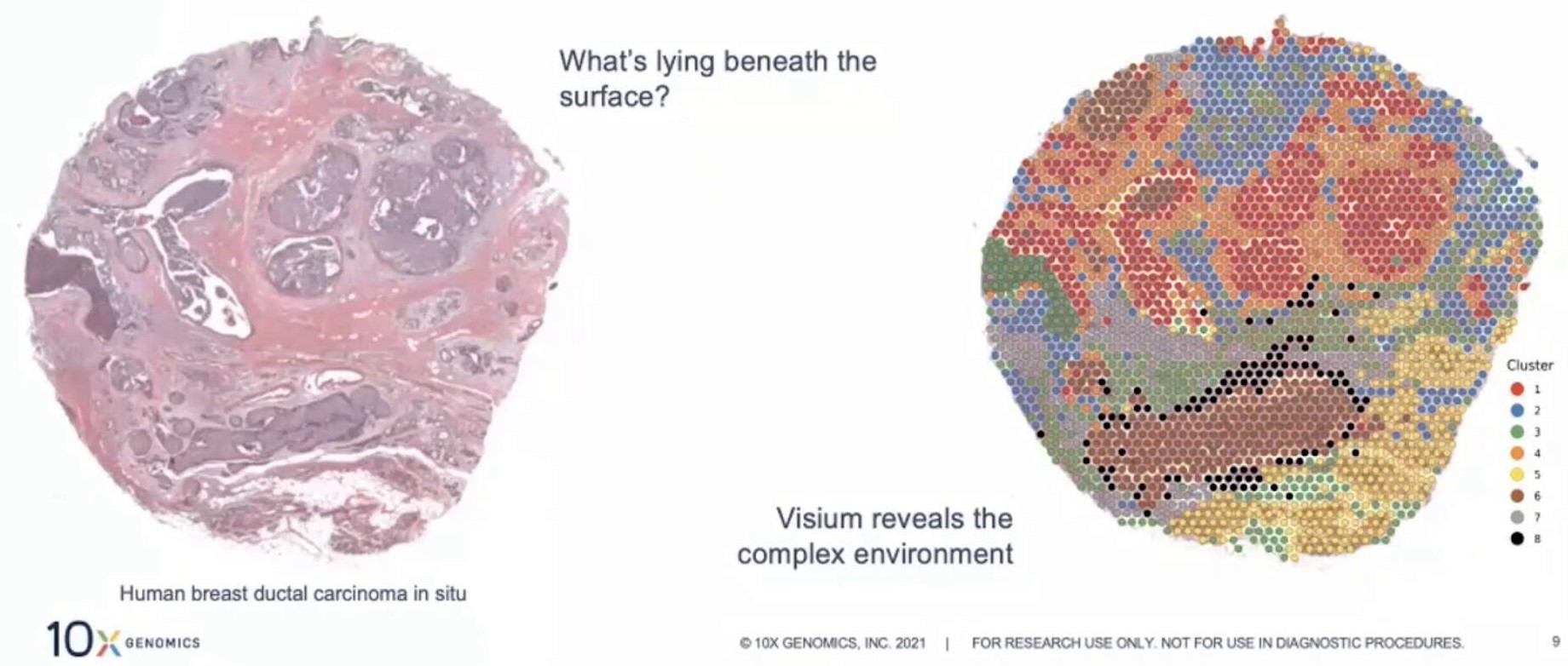
Spatial Transcriptomics
- Cells are held in place \(\implies\) transcripts identified within a region
- The current hot area in transcriptomics \(\implies\) Nature Methods “Method of the Year, 2020”
- Single-cell resolution is arguably here
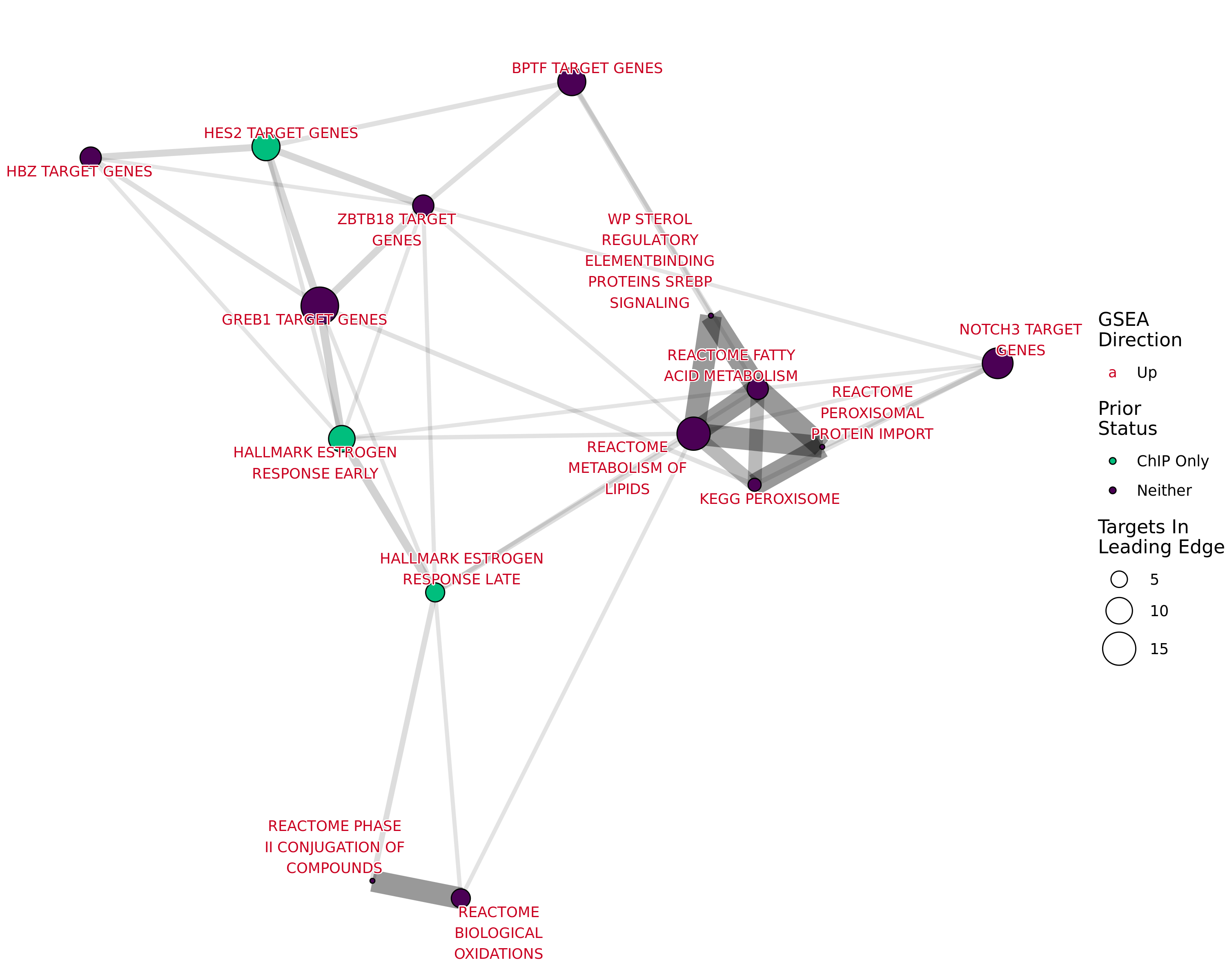
Pathway & Functional Analysis
- Look for biologically relevant signals in DE genes or network modules
- Enriched pathways
- Common transcription factors
- Drug target signals
- Compare to public datasets
(if appropriate)
- Interpretation is key
- Researchers love to invent stories
The Central Dogma
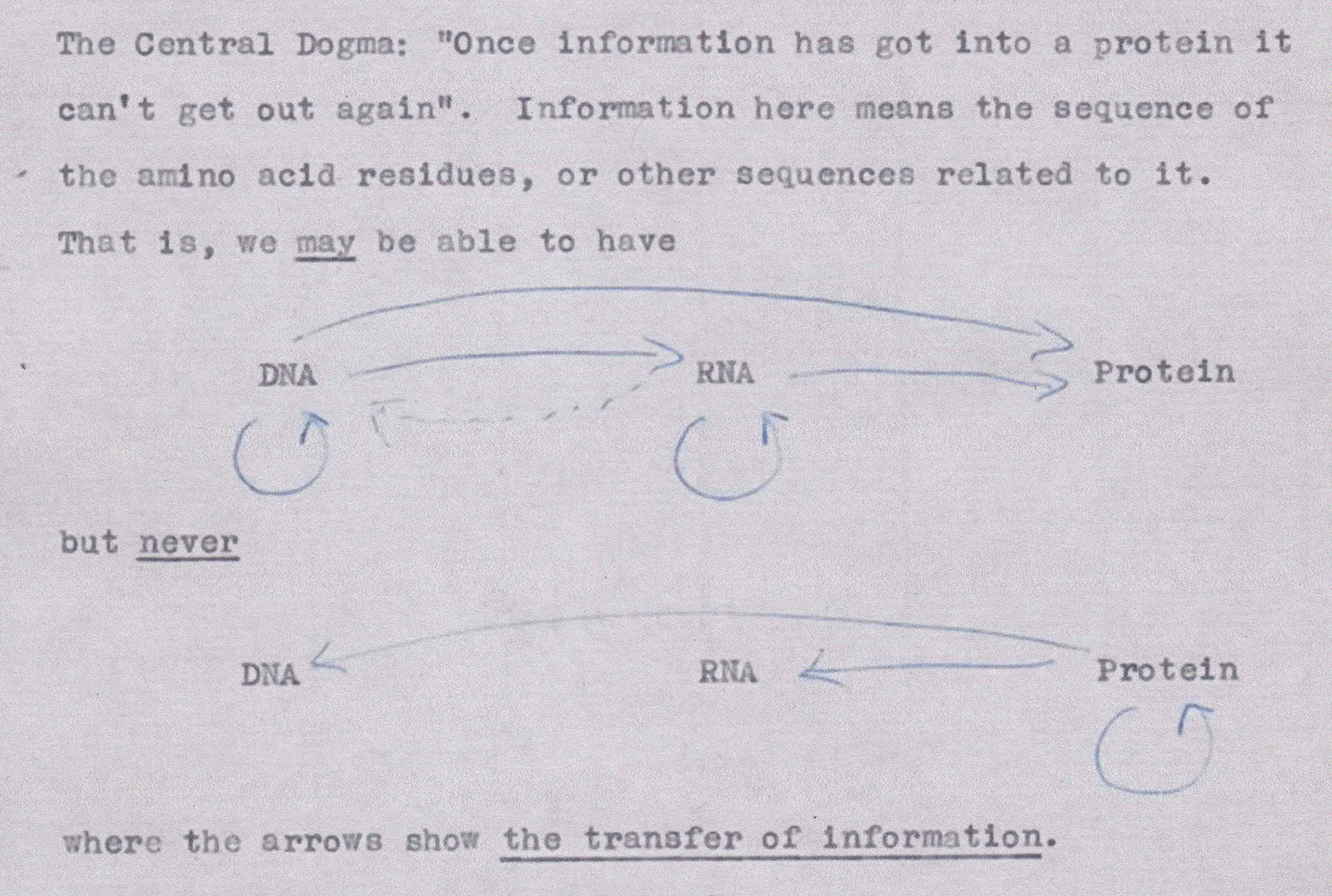
From Frances Crick, Ideas on Protein Synthesis, Unpublished Note, Wellcome Library, 1956
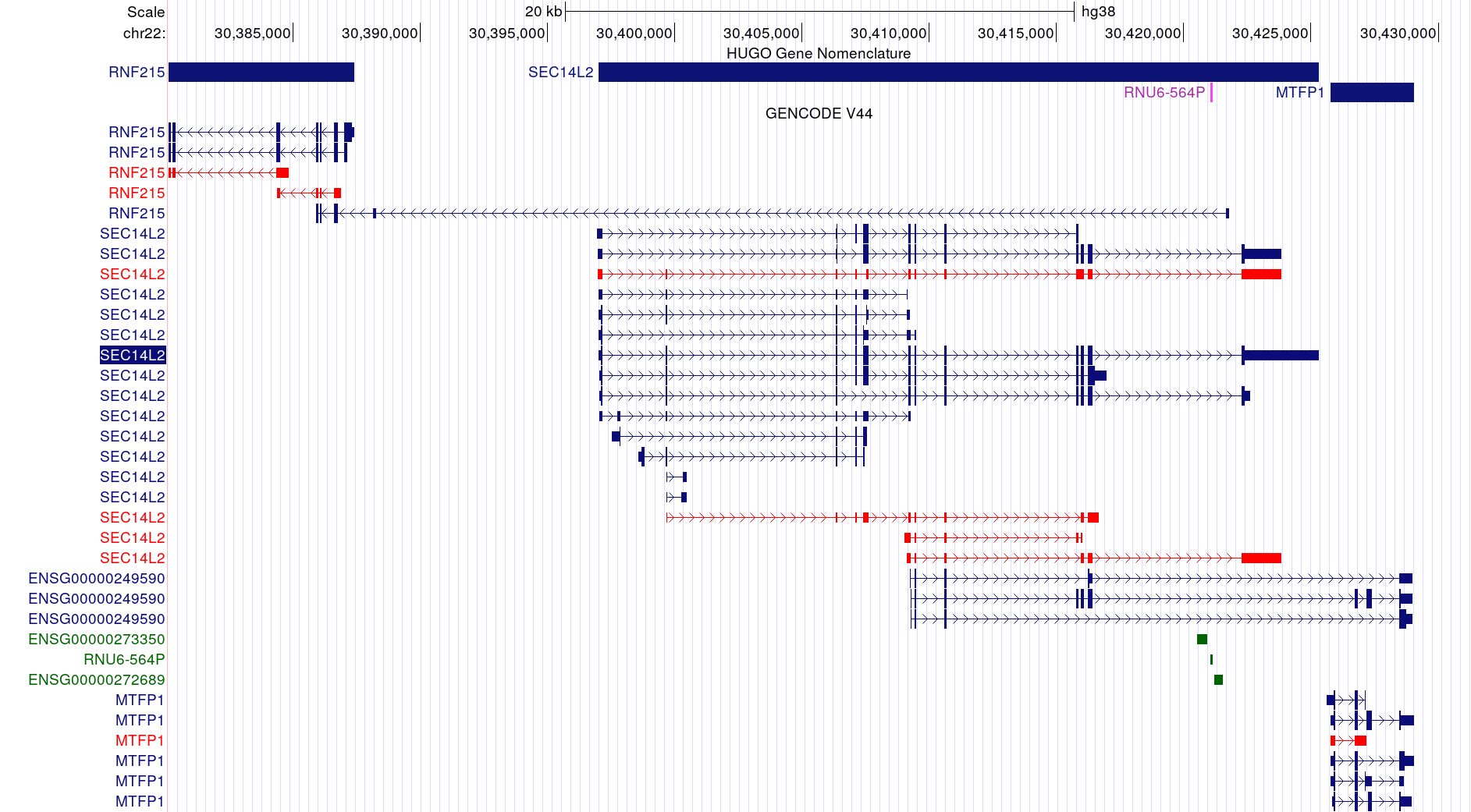
SEC14L2: A Random Example
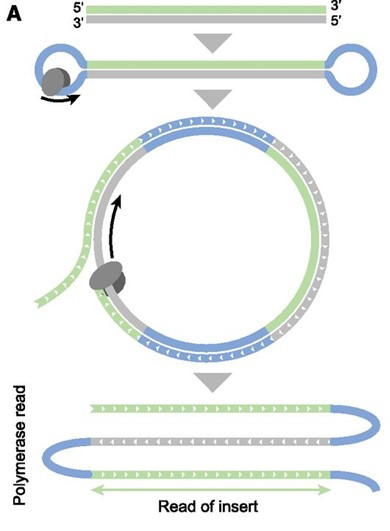
Long Reads
- Short read technology has ⇩⇩⇩ error rates
- Long reads have ⇧⇧⇧ error rates
- Reads are circularised
\(\implies\) errors corrected by repeat reads
- Great for identification of novel transcripts
\(\implies\) difficult to quantify- Creation of a custom reference transcriptome
- Challenging to refer back to functional annotations in reference
![]()